



Болезнь Фабри — это один из видов лизосомальных болезней накопления, который называется сфинголипидозом. Она вызвана накоплением в тканях гликолипида. Это заболевание является причиной появления новообразований кожи, боли в конечностях, плохого зрения, повторяющихся приступов повышения температуры тела, почечной недостаточности и заболевания сердца. Болезнь Фабри возникает, когда родители передают дефектный ген, вызывающий это заболевание, своим детям.
Болезнь Фабри возникает, когда организму не хватает необходимых ферментов для расщепления гликолипида.
Симптомы включают появление новообразований на коже, проблемы с глазами, почечную недостаточность и заболевание сердца.
Диагноз ставится на основании результатов пренатального скринингового обследования, скринингового обследования новорожденных и других анализов крови.
Большинство детей с болезнью Фабри доживают до взрослого возраста.
Лечение предусматривает заместительную терапию ферментами.
Существуют различные типы наследственных заболеваний. При многих наследственных нарушениях обмена веществ для того, чтобы это нарушение произошло, как правило, необходимы две копии патологического гена, поэтому больной ребенок унаследовал патологический ген от обоих родителей. (Часто ни один из родителей не страдает этим заболеванием). Некоторые наследственные нарушения обмена веществ сцеплены с X-хромосомой. Это означает, что у мальчиков заболевание может быть вызвано наследованием только одной копии патологического гена. (См. также Общие сведения о наследственных нарушениях обмена веществ).
Сфинголипидозы возникают, когда у людей отсутствуют ферменты, необходимые для расщепления (преобразования) сфинголипидов — веществ, защищающих клеточную поверхность и выполняющих определенные функции в клетках. Кроме болезни Фабри существует множество видов сфинголипидозов:
Болезнь Гоше (наиболее распространенная)
Болезнь Ниманна-Пика (типы А и В)
При болезни Фабри в тканях накапливается гликолипид, который является продуктом обмена жиров. Фермент, необходимый для расщепления гликолипида (альфа-галактозидаза А), не работает надлежащим образом. Так как дефектный ген этого редкого наследственного заболевания находится на Х-хромосоме, полная форма заболевания встречается только у мальчиков (см. Х-сцепленное наследование). Так как у девочек имеется две Х-хромосомы, у больных девочек могут присутствовать симптомы, но полная форма болезни Фабри не развивается.

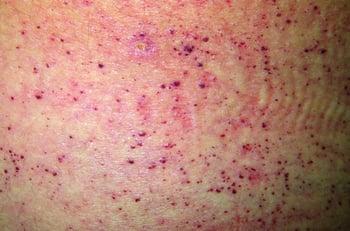
Накопление гликолипида вызывает нераковые (доброкачественные) новообразования кожи (ангиокератомы), образующиеся на нижней части туловища. Происходит помутнение роговицы, что приводит к ухудшению зрения. Может развиваться жгучая боль в руках и ногах, а у детей могут встречаться эпизоды повышения температуры. У детей с болезнью Фабри со временем развивается почечная недостаточность и заболевание сердца, хотя чаще всего они доживают до взрослого возраста. Почечная недостаточность может привести к повышению артериального давления, которое может закончиться инсультом.
Диагностика болезни Фабри
Пренатальные скрининговые обследования
Скрининговые обследования новорожденных
Другие анализы крови
До рождения болезнь Фабри у плода можно диагностировать с помощью пренатальных скрининговых обследований — биопсии ворсин хориона или амниоцентеза.
После рождения болезнь Фабри в некоторых штатах можно диагностировать при стандартном скрининговом обследовании новорожденных. Врачи измеряют уровень альфа-галактозидазы А в крови или в лейкоцитах.
Лечение болезни Фабри
Заместительная терапия ферментами
Иногда трансплантация почки
Врачи лечат оба заболевания с помощью заместительной терапии ферментами (агалсидазой бета). Лечение также заключается в приеме анальгетиков для облегчения боли и снижения температуры или противосудорожных препаратов.
При почечной недостаточности может потребоваться трансплантация почки.
Дополнительная информация
Ниже приведены некоторые ресурсы на английском языке, которые могут быть полезными. Обратите внимание, что составители СПРАВОЧНИКА не несут ответственности за содержание этих ресурсов.
Национальная организация по редким заболеваниям (National Organization for Rare Disorders, NORD): этот ресурс предоставляет родителям и семьям информацию о редких заболеваниях, включая список редких заболеваний, группы поддержки и ресурсы клинических исследований.
Информационный центр по генетическим и редким заболеваниям (Genetic and Rare Diseases Information Center, GARD): этот ресурс предоставляет и информацию о редких или генетических заболеваниях в доступной для понимания форме.




