




Dyslipidemia is elevation of plasma cholesterol, triglycerides (TGs), or both, or a low high-density lipoprotein cholesterol (HDL-C) level that contributes to the development of atherosclerosis. Causes may be primary (genetic) or secondary. Diagnosis is by measuring plasma levels of total cholesterol, TGs, and individual lipoproteins. Treatment involves dietary changes, exercise, and lipid-lowering drugs.
(See also Overview of Lipid Metabolism.)
Serum lipid levels are continuous; there is no precise threshold between normal and abnormal levels. A linear relation likely exists between lipid levels and cardiovascular risk (see table Cholesterol Levels and Cardiovascular Risk), so many people with “normal” cholesterol levels benefit from achieving still lower levels. Consequently, there are no numeric definitions of dyslipidemia; the term is applied to lipid levels for which treatment has proven beneficial.
Proof of benefit is strongest for lowering elevated low-density lipoprotein cholesterol (LDL-C) levels. In the general population, evidence is less strong for a benefit from lowering elevated TG and increasing low high-density lipoprotein cholesterol (HDL-C) levels.
HDL-C levels do not always predict cardiovascular risk. For example, high HDL-C levels caused by some genetic disorders may not be associated with a lower risk of cardiovascular disorders, and low HDL-C levels caused by some genetic disorders may not be associated with an increased risk of cardiovascular disorders. Although HDL-C levels predict cardiovascular risk in the general population, the increased risk may be caused by other factors, such as accompanying lipid and metabolic abnormalities, such as hypertriglyceridemia, rather than the HDL-C level itself.
Classification of Dyslipidemia
Dyslipidemias were traditionally classified by patterns of elevation in lipids and lipoproteins (Fredrickson phenotype—see table Lipoprotein Patterns). A more practical system categorizes dyslipidemias as primary or secondary and characterizes them by
Increases in cholesterol only: Pure or isolated hypercholesterolemia
Increases in TGs only: Pure or isolated hypertriglyceridemia
Increases in both cholesterol and TGs: Mixed or combined hyperlipidemias
This system does not take into account specific lipoprotein abnormalities (eg, low HDL-C or high LDL-C) that may contribute to disease despite normal cholesterol and TG levels.
Etiology of Dyslipidemia
Dyslipidemias may be
Primary: Genetic
Secondary: Caused by lifestyle and other factors
Both primary and secondary causes contribute to dyslipidemias in varying degrees. For example, in familial combined hyperlipidemia, expression may occur only in the presence of significant secondary causes.
Primary causes
Primary causes are single or multiple gene mutations that result either in overproduction or defective clearance of triglycerides and LDL or in underproduction or excessive clearance of HDL (see table Genetic (Primary) Dyslipidemias).
Secondary causes
Secondary causes contribute to many cases of dyslipidemia in adults.
The most important secondary cause of dyslipidemia in high-resource countries is
A sedentary lifestyle with excessive dietary intake of total calories, saturated fat, cholesterol, and trans fats
Trans fats are polyunsaturated or monounsaturated fatty acids to which hydrogen atoms have been added; they are used in some processed foods and are as atherogenic as saturated fat.
Other common secondary causes of dyslipidemia include
Primary biliary cirrhosis and other cholestatic liver diseases
estrogens cause a mixed effect (decrease LDL-C and increase HDL-C, but also increase TGs)
Secondary causes of low levels of HDL-C include cigarette smoking, anabolic steroids, HIV infection, and nephrotic syndrome.
Diabetes is an especially significant secondary cause because patients tend to have an atherogenic combination of high TGs; high small, dense LDL fractions; and low HDL (diabetic dyslipidemia, hypertriglyceridemic hyperapo B). Patients with type 2 diabetes are especially at risk. The combination may be a consequence of obesity, poor control of diabetes, or both, which may increase circulating free fatty acids (FFAs), leading to increased hepatic very-low-density lipoprotein (VLDL) production. TG-rich VLDL then transfers TG and cholesterol to LDL and HDL, promoting formation of TG-rich, small, dense LDL and clearance of TG-rich HDL. Diabetic dyslipidemia is often exacerbated by the increased caloric intake and physical inactivity that characterize the lifestyle of some patients with type 2 diabetes. Women with diabetes may be at special risk of cardiac disease as a result of this form of dyslipidemia.
Symptoms and Signs of Dyslipidemia
Dyslipidemia itself usually causes no symptoms, although very high TG levels can cause paresthesias, dyspnea, and confusion. Lipid disorders can lead to symptomatic end-organ disease, including
Vascular disease (eg, coronary artery disease (CAD), stroke, and peripheral arterial disease)
Acute pancreatitis can be caused be high TG levels (> 500 mg/dL [> 5.65 mmol/L])
Hepatosplenomegaly can be caused by very high TG levels
Findings in patients with severe dyslipidemia may include localized lipid deposits (xanthomas) or other findings caused by high serum concentrations or accumulation of lipids.
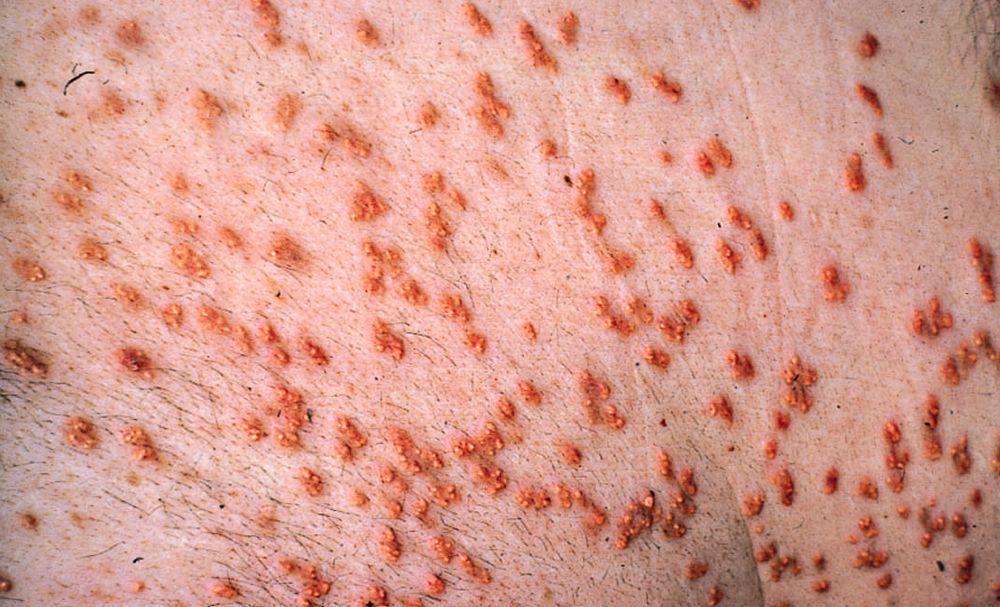
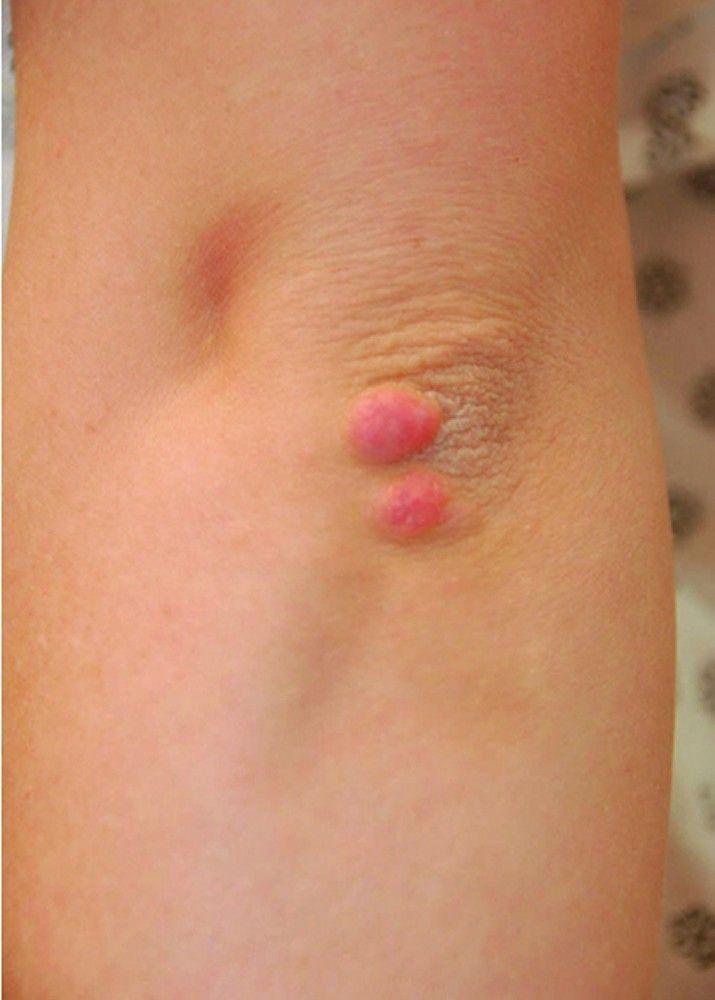
High LDL-C levels can cause tendinous xanthomas at the Achilles, elbow, and knee tendons and over metacarpophalangeal joints. Other clinical findings that occur in patients with high LDL-C (eg, in familial hypercholesterolemia or dysbetalipoproteinemia) include planar or tuberous xanthomas. Planar xanthomas are flat or slightly raised yellowish patches. Tuberous xanthomas are painless, firm nodules typically located over extensor surfaces of joints.
Patients with high levels of LDL-C can develop arcus corneae (lipid deposits in the cornea around the iris) and xanthelasma (lipid-rich, yellow plaques on the medial eyelids). Xanthelasma can also occur in patients with primary biliary cirrhosis and normal lipid levels.
Extremely high total cholesterol levels give a lactescent (milky) appearance to blood plasma.
Severe elevations of TGs can cause eruptive xanthomas over the trunk, back, elbows, buttocks, knees, hands, and feet. Severe hypertriglyceridemia (> 2000 mg/dL [> 22.6 mmol/L]) can also give retinal arteries and veins a creamy white appearance (lipemia retinalis).

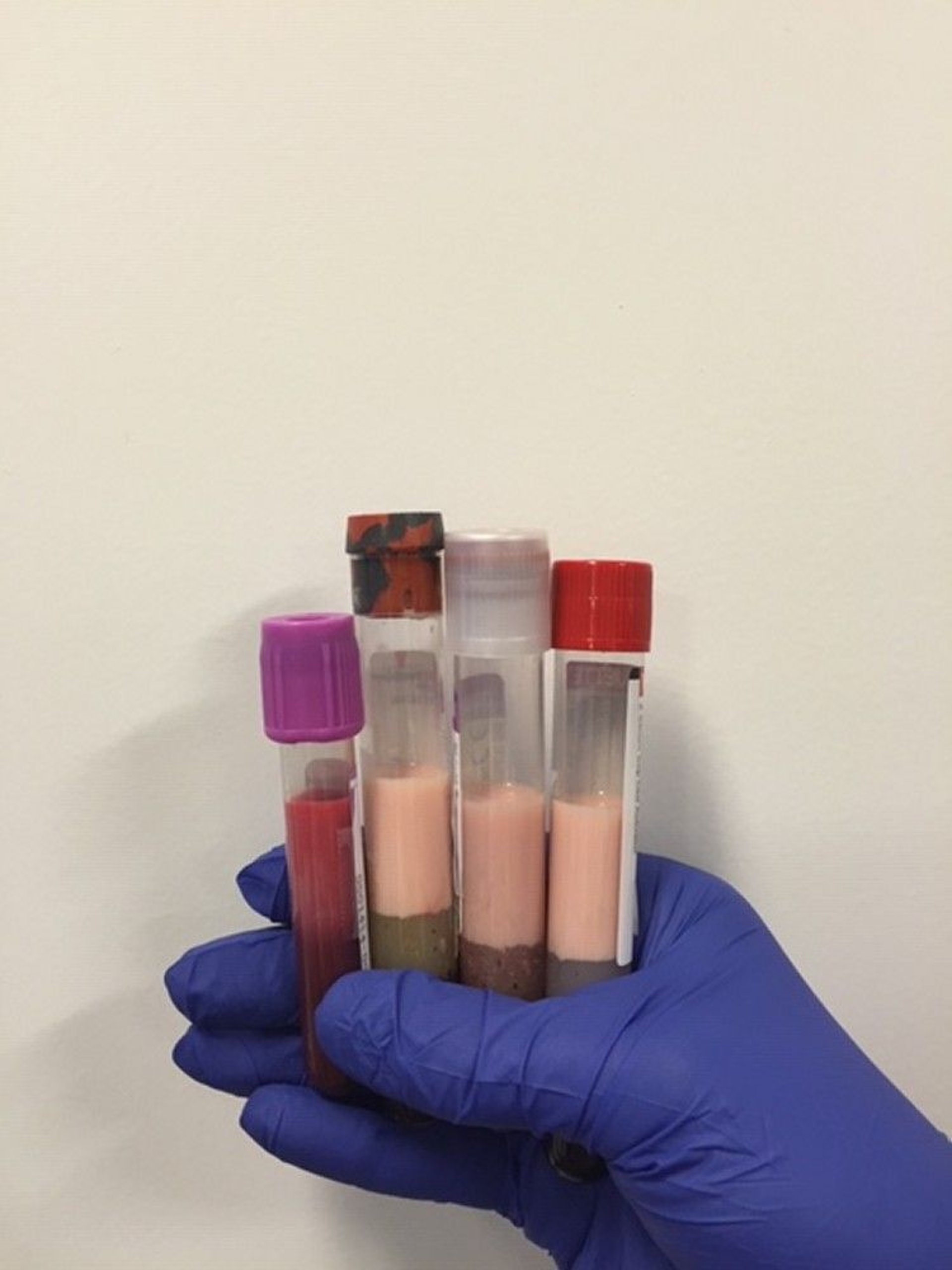
Image courtesy of Michael H. Davidson, MD.
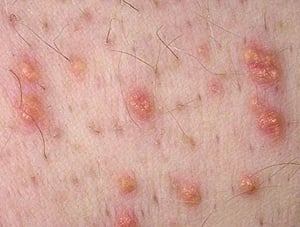
Photo provided by Thomas Habif, MD.
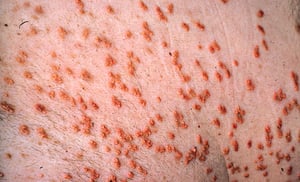
© Springer Science+Business Media
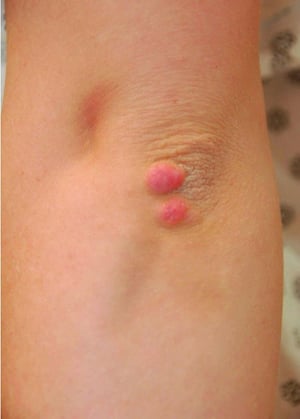
© Springer Science+Business Media
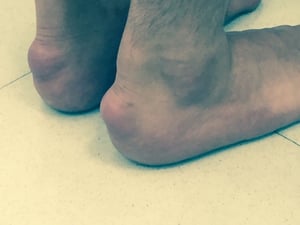
Image courtesy of Michael H. Davidson, MD.
Image courtesy of Michael H. Davidson, MD.

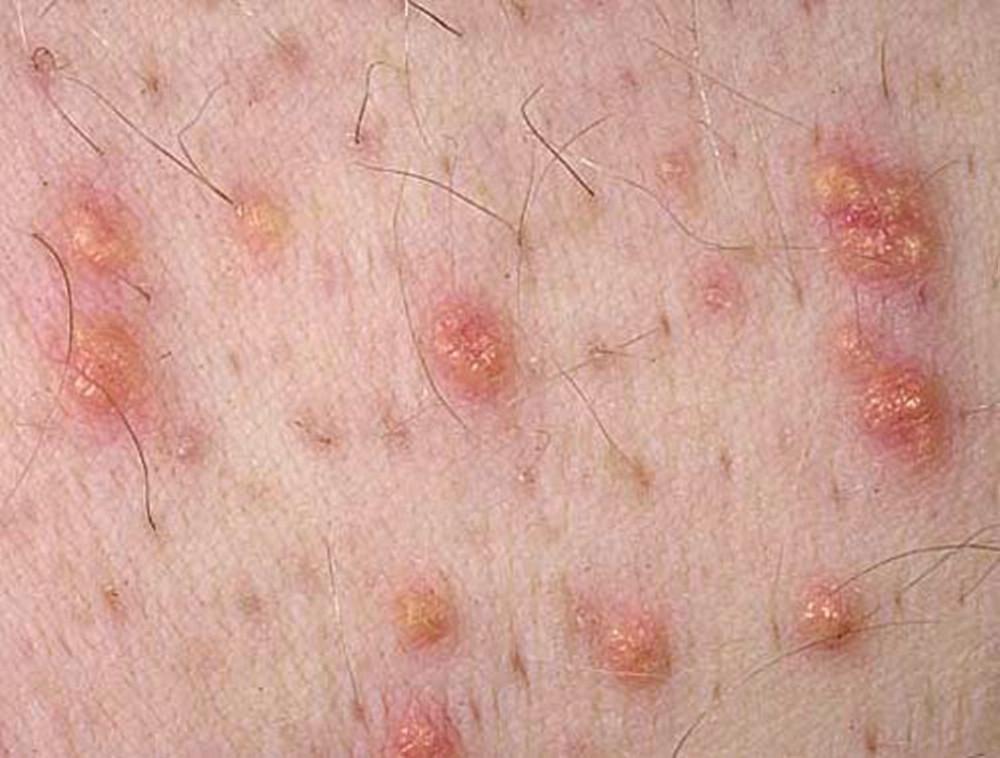
Photo provided by Thomas Habif, MD.

© Springer Science+Business Media

© Springer Science+Business Media

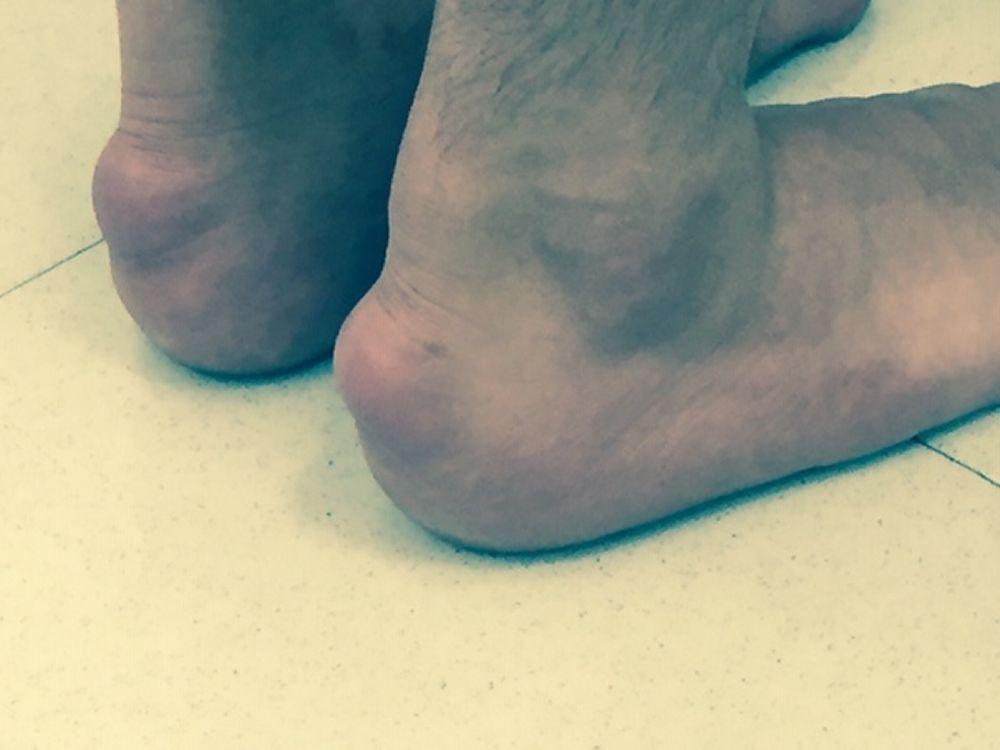
Image courtesy of Michael H. Davidson, MD.
Image courtesy of Michael H. Davidson, MD.
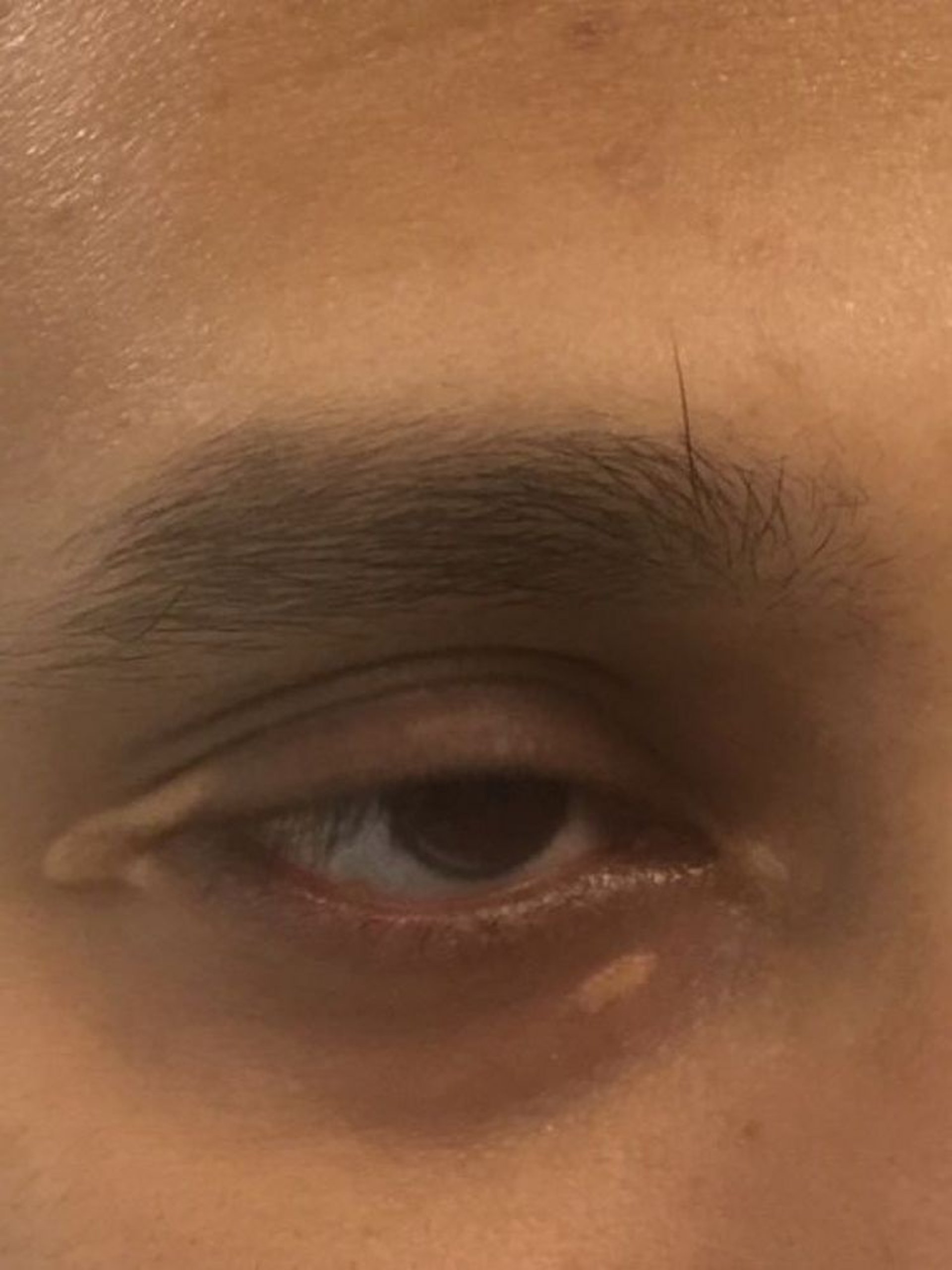
Image courtesy of Michael H. Davidson, MD.

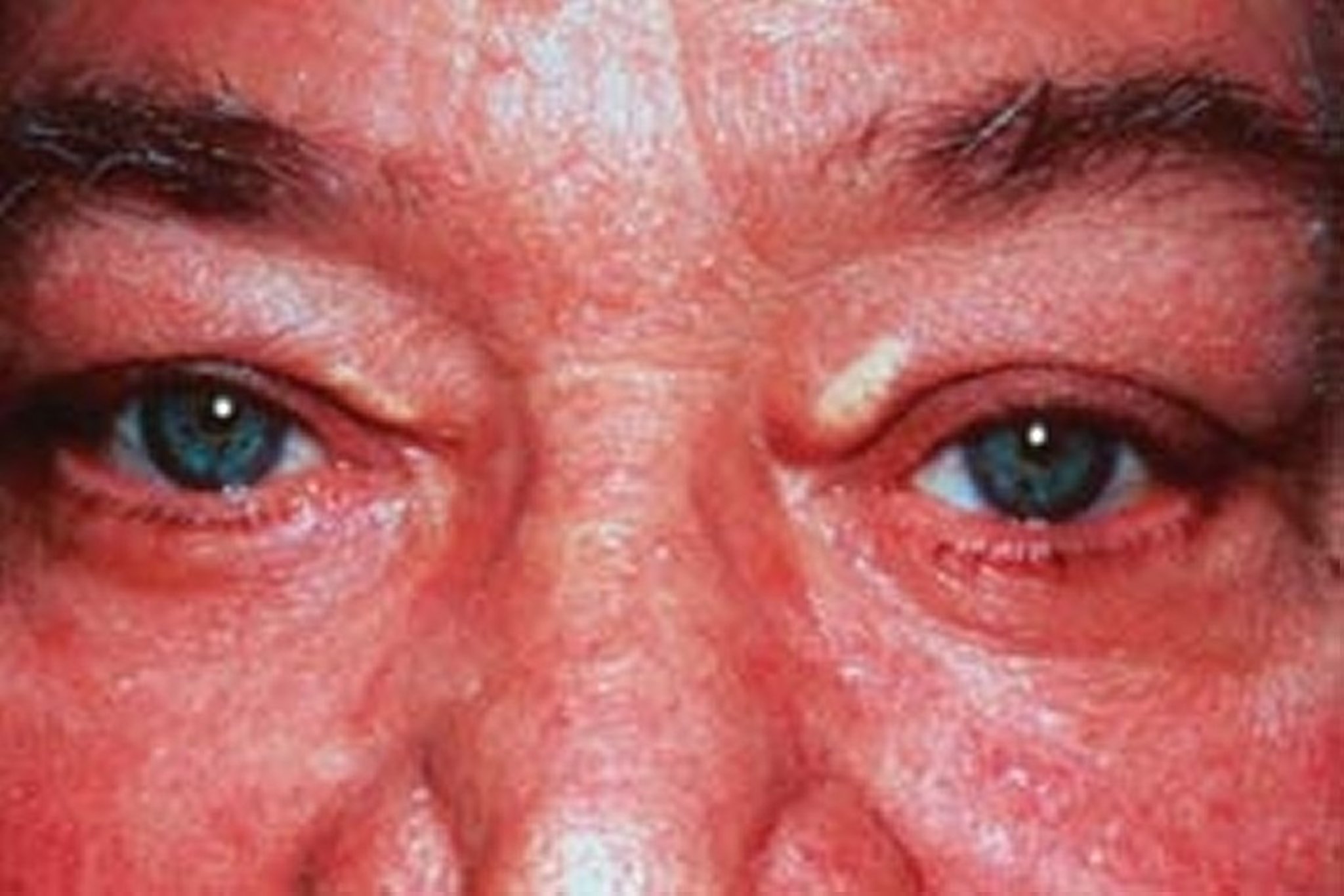
© Springer Science+Business Media
Diagnosis of Dyslipidemia
Serum lipid profile (measured total cholesterol, TG, and HDL-C and calculated LDL-C and VLDL-C)
Dyslipidemia is diagnosed by measuring serum lipids. Routine measurements (lipid profile) include total cholesterol (TC), TGs, HDL-C, and LDL-C; these results are used to calculate LDL-C and VLDL-C.
Dyslipidemia is often diagnosed with routine screening tests. It may also be suspected in patients with complications of dyslipidemia (eg, atherosclerotic disease). Physical findings are less common, and suggest primary dyslipidemia.
Primary lipid disorders are suspected when patients have
Physical signs of dyslipidemia, such as tendon xanthomas, which are pathognomonic for familial hypercholesterolemia
Onset of premature atherosclerotic disease (men < 55 years, women < 60 years)
Family history of premature atherosclerotic disease or severe hyperlipidemia
Serum cholesterol > 190 mg/dL (> 4.9 mmol/L)
Lipid profile measurement
Total cholesterol (TC), triglycerides (TGs), and HDL-C are measured directly. TC and TG values reflect cholesterol and TGs in all circulating lipoproteins, including chylomicrons, VLDL, intermediate-density lipoprotein (IDL), LDL, and HDL. TC values can vary by 10% and TGs by up to 25% day-to-day even in the absence of a disorder.
TC and HDL-C may be measured in the nonfasting state, but most patients should have all lipids measured while fasting (usually for 12 hours) for maximum accuracy and consistency.
Pearls & Pitfalls
|
Testing should be postponed if a patient has an acute illness, because TG and lipoprotein(a) levels increase and cholesterol levels decrease in inflammatory states. Also, lipid profiles can vary for about 30 days after an acute myocardial infarction (MI); however, results obtained within 24 hours after MI are usually reliable enough to guide initial lipid-lowering therapy.
LDL-C values are most often calculated as the amount of cholesterol not contained in HDL and VLDL. VLDL is estimated by TG ÷ 5 because the cholesterol concentration in VLDL particles is usually one fifth of the total lipid in the particle. Thus, the Friedewald formula estimates LDL-C as follows:

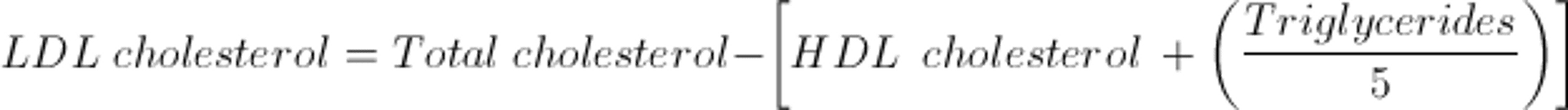
The calculated LDL cholesterol value incorporates measures of all non-HDL and nonchylomicron cholesterol, including that in IDL and lipoprotein (a) (Lp(a)).
For patients who have elevated TG levels, errors in estimating VLDL-C are magnified while using the constant factor of 5. The Martin-Hopkins equation may be used to obtain a more reliable estimate of LDL-C. The constant factor of 5 is replaced with a novel factor based on a patient's non-HDL-C and TG values (1). The novel factor is an adjustable factor that is based on the patient's non-HDL-C and TG levels and is derived from a table. Both the Friedewald and Martin-Hopkins equations were developed and validated for fasting patients with serum TG levels < 400 mg/dL (< 4.5 mmol/L).
The Martin-Hopkins equation is as follows:

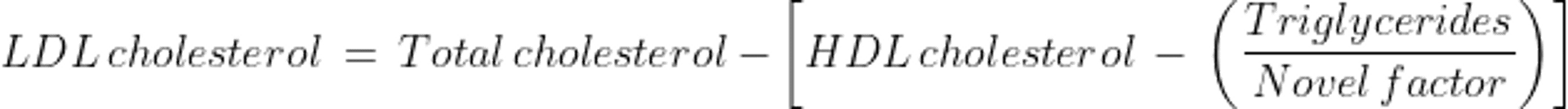
LDL-C can also be measured directly using plasma ultracentrifugation, which separates chylomicrons and VLDL fractions from HDL-C and LDL-C, and by an immunoassay method. Direct measurement may be useful in some patients with elevated TGs, but these direct measurements are not usually necessary.
Other tests
In some patients, additional lipid tests should be done.
Lp(a) levels and C-reactive protein levels should be measured in patients with premature atherosclerotic cardiovascular disease, cardiovascular disease (even if they have lower risk lipid levels; see table Cholesterol Levels and Cardiovascular Risk), or high LDL-C levels refractory to drug therapy. Lp(a) levels may also be directly measured in patients with borderline-high LDL-C levels to determine whether drug therapy is warranted.
Measurements of LDL particle number or apoprotein B-100 (apo B) is useful in patients with elevated TGs and the metabolic syndrome. Apo B provides similar information to LDL particle number because there is one apo B molecule for each LDL particle. Apo B measurement includes all atherogenic particles, including remnants and Lp(a).
Apo B value reflects all non-HDL-C (in VLDL, IDL, and LDL) and is more predictive of CAD risk than LDL-C. Non-HDL-C (TC − HDL-C) is also more predictive of CAD risk than LDL-C, especially in patients with hypertriglyceridemia.
Secondary causes
Tests for secondary causes of dyslipidemia should be done in most patients with newly diagnosed dyslipidemia and repeated when a component of the lipid profile has inexplicably changed for the worse. Such tests include measurements of
Creatinine
Fasting glucose and/or glycosylated hemoglobin (HbA1C)
Liver enzymes
Thyroid-stimulating hormone (TSH)
Urinary protein
Diagnosis reference
1. Martin SS, Blaha MJ, Elshazly MB, et al: Comparison of a novel method vs the Friedewald equation for estimating low-density lipoprotein cholesterol levels from the standard lipid profile. JAMA 310(19):2061-2068, 2013. doi:10.1001/jama.2013.280532
Screening for Dyslipidemia
Screening is done using a fasting lipid profile (TC, TGs, HDL-C, and calculated LDL-C). Medical society guidelines vary regarding when to begin screening. Based on the risk factors, screening can begin as early as age 2 in children with family history of heart disease or familial hypercholesterolemia.
Lipid measurement should be accompanied by assessment for other cardiovascular risk factors, including
Cigarette use
Family history of CAD in a male first-degree relative before age 55 years or a female first-degree relative before age 65 years
Screening in children
Most physicians recommend screening per the 2012 National Heart Lung and Blood Institute Guidelines (1) as follows
Children with risk factors (eg, diabetes, hypertension, family history of severe hyperlipidemia or premature CAD): Fasting lipid profile once at age 2 to 8
Children with no risk factors: Non-fasting or fasting lipid profile once before puberty (usually age 9 to 11) and once more at age 17 to 21
Screening in adults
Adults should be screened at age 20 (2, 3) and every 5 years thereafter.
An age to discontinue screening has not been established, but evidence supports screening of patients into their 80s, especially if they have atherosclerotic cardiovascular disease (4).
Patients with an extensive family history of heart disease—heart attack, stroke, or coronary artery disease before age 55 (in men) or age 65 (in women) without known risk factors, such as high LDL, smoking, diabetes, or obesity, or known family history of high Lp(a)—should also be screened by measuring Lp(a) levels.
Screening references
1. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents. 2011.
2. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al: 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk. J Am Coll Cardiol 63:2935–2959, 2014. doi: 10.1016/j.jacc.2013.11.005
3. Grundy SM, Stone NJ, Bailey AL, et al: 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139: e1082–e1143, 2019. doi: 10.1161/CIR.0000000000000625
4. Kleipool EE, Dorresteijn JA, Smulders YM, et al: Treatment of hypercholesterolaemia in older adults calls for a patient-centred approach. Heart 106(4):261-266, 2020. doi:10.1136/heartjnl-2019-315600
Treatment of Dyslipidemia
Lifestyle changes (eg, exercise, dietary modification)
For high TG, fibrates, omega-3 fatty acids, and sometimes other measures
General principles
The main goal for dyslipidemia treatment is prevention of atherosclerotic cardiovascular disease (ASCVD), including acute coronary syndromes, stroke, transient ischemic attack, or peripheral arterial disease presumed caused by atherosclerosis. Treatment is indicated for all patients with ASCVD (secondary prevention) and for some without (primary prevention).
Treatment of children is controversial because there is no evidence that lowering lipid levels in childhood effectively prevents heart disease in adulthood. Moreover, the safety and effectiveness of long-term lipid-lowering treatment in children are unknown. Nevertheless, the American Academy of Pediatrics (AAP) recommends treatment for some children who have elevated LDL-C levels. Children with heterozygous familial hypercholesterolemia should be treated beginning at age 8 to 10 years. Children with homozygous familial hypercholesterolemia require diet, medications, and often LDL apheresis to prevent premature death; treatment is begun when the diagnosis is made.
Treatment options depend on the specific lipid abnormality, although different lipid abnormalities often coexist. In some patients, a single abnormality may require several therapies; in others, a single treatment may be adequate for several abnormalities. Treatment should always include treatment of hypertension and diabetes and smoking cessation. Treatment should also include daily low-dose aspirin in patients age 40 to 79 years with a low risk of bleeding and with a 10-year risk of myocardial infarction or death due to CAD of ≥ 20%. In general, treatment options for men and women are the same.
Elevated LDL-C treatment
For all individuals, the prevention of ASCVD requires an emphasis on a heart-healthy lifestyle, particularly diet and exercise. Other options to lower LDL-C in all age groups include medications, dietary supplements, and procedural interventions. Many of these options are also effective for treating other lipid abnormalities.
Dietary changes help to maintain ideal body weight and provide other benefits. These changes include
Decreasing intake of saturated fats and cholesterol
Increasing the proportion of dietary fiber and complex carbohydrates
Referral to a dietitian is often useful.
Exercise lowers LDL-C in some people and also helps maintain ideal body weight.
Dietary changes and exercise should be used whenever feasible, but American Heart Association (AHA)/American College of Cardiology (ACC) guidelines recommend also using drug therapy for certain groups of patients after discussion of the risks and benefits of statin therapy (1).
For drug therapy in adults, the AHA/ACC Guidelines recommend treatment with a statin for 4 groups of patients, comprised of those with any of the following:
Clinical ASCVD
LDL-C ≥ 190 mg/dL (≥ 4.9 mmol/L)
Age 40 to 75 years, with diabetes and LDL-C 70 to 189 mg/dL (1.8 to 4.9 mmol/L)
Age 40 to 75 years, with LDL-C 70 to 189 mg/dL (1.8 to 4.9 mmol/L), and an estimated 10-year risk of ASCVD ≥ 7.5%
Risk of ASCVD is estimated using the pooled cohort risk assessment equations. This risk calculator is based on sex, age, race, total and HDL-C, systolic and diastolic blood pressure, diabetes and smoking status, and use of antihypertensives or statins.
Increased lifetime risk (identified using the AHA/ACC risk calculator) is relevant because 10-year risk may be low in younger patients, in whom longer-term risk should be taken into account.
When considering whether to give a statin, clinicians may also take into account other factors, including
LDL-C ≥ 160 mg/dL (4.1 mmol/L)
Family history of premature ASCVD (ie, age of onset < 55 years in a male first-degree relative, or < 65 years in a female first-degree relative)
High-sensitivity C-reactive protein ≥ 2 mg/L (≥ 19 nmol/L)
Coronary artery calcium score ≥ 300 Agatston units (or ≥ 75th percentile for the patient's demographic)
Ankle-brachial index < 0.9
Increased lifetime risk
Statins are the treatment of choice for LDL-C reduction because evidence has demonstrated that they reduce cardiovascular morbidity and mortality (2). Other classes of lipid-lowering drugs are not the first choice for treating elevated LDL-C because they have not demonstrated equivalent efficacy for decreasing ASCVD.
Statins inhibit hydroxymethylglutaryl CoA reductase, a key enzyme in cholesterol synthesis, leading to up-regulation of LDL receptors and increased LDL clearance. They reduce LDL-C by up to 60% and produce small increases in HDL-C and modest decreases in TGs. Statins also appear to decrease intra-arterial inflammation, systemic inflammation, or both by stimulating production of endothelial nitric oxide and may have other beneficial effects.
Statin therapy is classified as high-, moderate-, or low-intensity and is given based on treatment group and age (see table Statins for ASCVD Prevention). The choice of statin depends on the patient's co-morbidities, other drugs, risk factors for adverse events, statin intolerance, cost, and patient preference.
Adverse effects with statins are uncommon but include liver enzyme elevations and myositis or rhabdomyolysis
In patients with ASCVD, risk reduction increases with decreasing LDL-C levels. Thus, initial treatment is a statin at maximally tolerated dose with the goal of lowering LDL-C by > 50% (high-intensity therapy). For very high-risk patients with ASCVD (eg, those with a recent myocardial infarction or unstable angina or with high-risk comorbidities such as diabetes3, 4).
Non-statin medications also have a place in lowering LDL-C (see table Non-Statin Lipid–Lowering Drugs). The 2022 ACC Expert Consensus Decision Pathway on the Role of Nonstatin Therapies provides guidance to clinicians on the role of non-statin therapy in both primary and secondary prevention of ASCVD (5). Statins are generally first-line medications, but in patients with statin intolerance, or those who are not able to achieve target LDL-C levels despite statin use, non-statin medications are used.
The adenosine triphosphate citrate lyase inhibitor,6, 7
Bile acid sequestrantsnicotinic acid
The cholesterol absorption inhibitor
PCSK9 monoclonal antibodies3).
SiRNA targeting PCSK9
Dietary supplements that lower LDL-C levels include fiber supplements and commercially available margarines and other products containing plant sterols (sitosterol, campesterol) or stanols. Fiber supplements decrease cholesterol levels in multiple ways, including decreased absorption and increased excretion. Oat-based fiber supplements can decrease total cholesterol by up to 18%. Plant sterols and stanols decrease cholesterol absorption by displacing cholesterol from intestinal micelles and can reduce LDL-C by up to 10% without affecting HDL-C or TGs.
Cholesterylester transfer protein inhibitors are a class of drugs that are under investigation and that may simultaneously raise HDL-C and lower LDL-C. Cholesterylester transfer protein (CETP) is a plasma glycoprotein produced in the liver and adipose tissue that circulates in the blood bound primarily to HDL, which mediates the transfer of cholesteryl esters from HDL to ApoB containing particles.
Medications for homozygous familial hypercholesterolemia
Procedural approaches
Procedural approaches are reserved for patients with severe hyperlipidemia (LDL-C > 300 mg/dL [> 7.74 mmol/L]) and no vascular disease. LDL apheresis may be done in patients with LDL-C > 200 mg/dL (> 5.16 mmol/L) and vascular disease that is refractory to conventional therapy, such as occurs with familial hypercholesterolemia. Options include LDL apheresis (in which LDL is removed by extracorporeal plasma exchange) and, rarely, ileal bypass (to block reabsorption of bile acids) and liver transplantation (which transplants LDL receptors). LDL apheresis is the procedure of choice in most instances when maximally tolerated therapy fails to lower LDL-C adequately. Apheresis is also the usual therapy in patients with the homozygous form of familial hypercholesterolemia who have limited or no response to drug therapy.
Elevated LDL-C in children
Childhood risk factors besides family history and diabetes include cigarette smoking, hypertension, low HDL-C (< 35 mg/dL [< 0.9 mmol/L]), obesity, and physical inactivity.
The American Heart Association recommends dietary treatment for children with LDL-C > 110 mg/dL (> 2.8 mmol/L) (8).
Drug therapy is recommended for children > 8 years and with either of the following:
Poor response to dietary therapy, LDL-C ≥ 190 mg/dL (≥ 4.9 mmol/L), and no family history of premature cardiovascular disease
LDL-C ≥ 160 mg/dL (≥ 4.13 mmol/L) and a family history of premature cardiovascular disease or ≥ 2 risk factors for premature cardiovascular disease
Medications used in children include many of the statins. Children with familial hypercholesterolemia may require a second drug to achieve LDL-C reduction of at least 50%.
Elevated triglycerides
Although it is unclear whether elevated TGs independently contribute to cardiovascular disease, they are associated with multiple metabolic abnormalities that contribute to coronary artery disease (eg, diabetes, metabolic syndrome). Consensus is emerging that lowering elevated TGs is beneficial. No target goals exist, but levels < 150 mg/dL (< 1.7 mmol/L) are generally considered desirable. No guidelines specifically address treatment of elevated TGs in children.
The overall treatment strategy is to first implement lifestyle changes, including exercise, weight loss, and avoidance of concentrated dietary sugar and alcohol. Intake of 2 to 4 servings/week of marine fish high in omega-3 fatty acids may be effective, but the amount of omega-3 fatty acids is often lower than needed; supplemental doses may be helpful. In patients with diabetes, glucose levels should be tightly controlled. If these measures are ineffective, lipid-lowering drugs should be considered. Patients with very high TG levels (> 1,000 mg/dL [> 11 mmol/L]) may need to begin drug therapy at diagnosis to more quickly reduce the risk of acute pancreatitis.
Fibrates reduce TGs by about 50%. They appear to stimulate endothelial lipoprotein lipase (LPL), leading to increased fatty acid oxidation in the liver and muscle and decreased hepatic VLDL synthesis. They also increase HDL-C by up to 20%. Fibrates can cause gastrointestinal adverse effects, including dyspepsia, abdominal pain, and elevated liver enzymes. They uncommonly cause cholelithiasis
Statins can be used in patients with TGs < 500 mg/dL (< 5.65 mmol/L) if LDL-C elevations are also present; statins may reduce both LDL-C and TGs through reduction of VLDL. If only TGs are elevated, fibrates are the drug of choice.
Omega-3 fatty acids> 5.65 mmol/L).
The Apo CIII inhibitor (an antisense inhibitor of apo CIII), volanesorsen, is available in some countries. It lowers triglyceride levels in patients with severely elevated TG levels, including people with lipoprotein lipase deficiency. It is given as a weekly injection.
Low HDL-C
Although higher HDL-C levels predict lower cardiovascular risk, it is not clear whether treatments to increase HDL-C levels decrease risk of death. Guidelines in the Third Report of the NCEP Expert Panel define low HDL-C as < 40 mg/dL [< 1.04 mmol/L]; the guidelines do not specify an HDL-C target level and recommend interventions to raise HDL-C only after LDL-C targets have been reached (9). Treatments for LDL-C and TG reduction often increase HDL-C, and the 3 objectives can sometimes be achieved simultaneously.
No guidelines specifically address treatment of low HDL-C in children.
Pearls & Pitfalls
|
Treatment includes lifestyle changes such as an increase in exercise and weight loss. Alcohol raises HDL-C but is not routinely recommended as a therapy because of its many other adverse effects. Drugs may be successful in raising levels when lifestyle changes alone are insufficient, but it is uncertain whether raising HDL-C levels reduces mortality.
insulin resistance, and hyperuricemia and gout. It may also increase homocysteine levelsischemic stroke.
Fibrates increase HDL-C. Fibrates may decrease cardiovascular risk in patients with TGs > 200 mg/dL (> 2.26 mmol/L) and HDL-C < 40 mg/dL (< 1.04 mmol/L).
Elevated Lp(a)
The usual approach in patients with elevated Lp(a) is to lower LDL-C aggressively. LDL apheresis has been used to lower Lp(a) in patients with high Lp(a) levels and progressive vascular disease.
Treatment in patients with comorbidities
Treatment of diabetic dyslipidemia should always involve lifestyle changes and statins to reduce LDL-C. To decrease the risk of pancreatitis, fibrates can be used to decrease TGs when levels are > 500 mg/dL (>
Treatment of dyslipidemia in patients with hypothyroidism, chronic kidney disease, liver disease, or a combination of these disorders involves treating the underlying disorders primarily and lipid abnormalities secondarily. Abnormal lipid levels in patients with low-normal thyroid function (high-normal TSH levels) improve with hormone replacement. Reducing the dosage of or stopping drugs that cause lipid abnormalities should be considered.
Monitoring treatment
Lipid levels should be monitored periodically after starting treatment. No data support specific monitoring intervals, but measuring lipid levels 2 to 3 months after starting or changing therapies and once or twice yearly after lipid levels are stabilized is common practice.
Liver and severe muscle toxicity with statin use occurs in 0.5 to 2% of all users. Routine monitoring of liver enzyme levels is not necessary, and routine measurement of creatine kinase (CK) is not useful to predict the onset of rhabdomyolysis. Muscle enzyme levels need not be checked regularly unless patients develop myalgias or other muscle symptoms. If statin-induced muscle damage is suspected, statin use is stopped and CK may be measured. When muscle symptoms subside, a lower dose or a different statin can be tried. If symptoms do not subside within 1 to 2 weeks of stopping the statin, another cause should be sought for the muscle symptoms (eg, polymyalgia rheumatica).
Treatment references
1. Grundy SM, Stone NJ, Bailey AL, et al: 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139: e1082–e1143, 2019. doi: 10.1161/CIR.0000000000000625
2. US Preventive Services Task Force, Mangione CM, Barry MJ, et al: Statin Use for the Primary Prevention of Cardiovascular Disease in Adults: US Preventive Services Task Force Recommendation Statement. JAMA 328(8):746-753, 2022. doi:10.1001/jama.2022.13044
3. Sabatine MS, Giugliano RP, Keech AC, et al: Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 376:1713–1722, 2017. 15
4. Schwartz GG, Steg PG, Szarek M, et al: Alirocumab and cardiovascular outcomes after acute coronary syndrome. New Engl J Med 379:2097–2107, 2018. doi: 10.1056/NEJMoa1801174.
5. Writing Committee, Lloyd-Jones DM, Morris PB, et al:. 2022 ACC Expert Consensus Decision Pathway on the Role of Nonstatin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 2023 Jan 3;81(1):104]. J Am Coll Cardiol 80(14):1366-1418, 2022. doi:10.1016/j.jacc.2022.07.006
6. Goldberg AC, Leiter LA, Stroes ESG, et al. Effect of Bempedoic Acid vs Placebo Added to Maximally Tolerated Statins on Low-Density Lipoprotein Cholesterol in Patients at High Risk for Cardiovascular Disease: The CLEAR Wisdom Randomized Clinical Trial [published correction appears in JAMA. 2020 Jan 21;323(3):282]. JAMA 2019;322(18):1780-1788. doi:10.1001/jama.2019.16585
7. Ray KK, Bays HE, Catapano AL, et al. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N Engl J Med 2019;380(11):1022-1032. doi:10.1056/NEJMoa1803917
8. de Ferranti SD, Steinberger J, Ameduri R, et al: Cardiovascular Risk Reduction in High-Risk Pediatric Patients: A Scientific Statement From the American Heart Association. Circulation 139(13):e603-e634, 2019. doi:10.1161/CIR.0000000000000618
9. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III): Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106(25):3143-3421, 2002.
Key Points
Elevated lipid levels are a risk factor for atherosclerosis and thus can lead to symptomatic coronary artery disease and peripheral arterial disease.
Causes of dyslipidemia include a sedentary lifestyle with excessive dietary intake of calories, saturated fat, cholesterol, and trans fats and/or genetic (familial) abnormalities of lipid metabolism.
Diagnose using serum lipid profile (measured total cholesterol, triglycerides, and high-density lipoprotein [HDL] cholesterol and calculated low-density lipoprotein [LDL] cholesterol and very low-density lipoprotein [VLDL] cholesterol).
Screening tests should be done at age 9 to 11 years and again at age 17 to 21 years (age 2 to 8 if there is a strong family history of severe hyperlipidemia or premature coronary artery disease or other risk factors); adults are screened every 5 years beginning at age 20.
Treatment with a statin is indicated to reduce risk of atherosclerotic cardiovascular disease for all patients in 4 major risk groups as defined by the American College of Cardiology/American Heart Association and for those without who have certain other combinations of risk factors and elevated lipid levels.






