




Polycystic kidney disease (PKD) is a hereditary disorder of renal cyst formation causing gradual enlargement of both kidneys, sometimes with progression to renal failure. Almost all forms are caused by a familial genetic mutation. Symptoms and signs include flank and abdominal pain, hematuria, and hypertension. Diagnosis is by CT or ultrasonography. Treatment is symptomatic before renal failure and with dialysis or transplantation afterward.
(See also Overview of Cystic Kidney Disease.)
Etiology of ADPKD
Inheritance of polycystic kidney disease (PKD) is
Autosomal dominant
Recessive
Sporadic (rare)
Autosomal dominant polycystic kidney disease (ADPKD) has an incidence of 1/1000 and accounts for about 5% of patients with end-stage kidney disease (ESKD) requiring renal replacement therapy. Clinical manifestations are rare before adulthood, but penetrance is essentially complete; all patients ≥ 80 years have some signs.
In contrast, autosomal recessive polycystic kidney disease is rare; incidence is 1/10,000. It frequently causes renal failure during childhood.
In 86 to 96% of cases, ADPKD is caused by mutations in the PKD1 gene on chromosome 16, which codes for the protein polycystin 1; most other cases are caused by mutations in the PKD2 gene on chromosome 4, which codes for polycystin 2. A few familial cases are unrelated to either locus.
Pathophysiology of ADPKD
Polycystin 1 may regulate tubular epithelial cell adhesion and differentiation; polycystin 2 may function as an ion channel, with mutations causing fluid secretion into cysts. Mutations in these proteins may alter the function of renal cilia, which enable tubular cells to sense flow rates. A leading hypothesis proposes that tubular cell proliferation and differentiation are linked to flow rate and that ciliary dysfunction may thus lead to cystic transformation.
Vasopressin promotes cell growth and fluid secretion via the cyclic AMP pathway, which leads to increase in the size and number of cysts in polycystic kidney disease.
Early in the disorder, tubules dilate and slowly fill with glomerular filtrate. Eventually, the tubules separate from the functioning nephron and fill with secreted rather than filtered fluid, forming cysts. Hemorrhage into cysts may occur, causing hematuria. Patients are also at higher risk of acute pyelonephritis, cyst infections, and urinary calculi (in 20%). Vascular sclerosis and interstitial fibrosis eventually develop via unknown mechanisms and typically affect < 10% of tubules; nonetheless, renal failure develops in about 35 to 45% of patients by age 60.
Extrarenal manifestations are common:
Hepatic cysts are present in most patients; these typically do not affect liver function.
Patients also have a higher incidence of pancreatic and intestinal cysts, colonic diverticula, and inguinal and abdominal wall hernias.
Valvular heart disorders (most often mitral valve prolapse and aortic regurgitation) can be detected by cardiac ultrasonography in 25 to 30% of patients; other valvular disorders may be due to collagen abnormalities.
Aortic regurgitation results from aortic root dilation due to arterial wall changes (including aortic aneurysm).
Coronary artery aneurysms occur.
Cerebral aneurysms are present in about 4% of young adults and up to 10% of older adults. Aneurysms rupture in 65 to 75% of patients, usually before age 50; risk factors include family history of aneurysm or rupture, larger aneurysms, and poorly controlled hypertension.
Symptoms and Signs of ADPKD
Autosomal dominant polycystic kidney disease usually causes no symptoms initially; one half of patients remain asymptomatic, never develop renal insufficiency or failure, and are never diagnosed. Most patients who develop symptoms do so by the end of their 20s.
Symptoms include low-grade flank, abdominal, and lower back pain due to cystic enlargement and symptoms of infection. Acute pain, when it occurs, is usually due to hemorrhage into cysts or passage of a calculus. Fever is common with acute pyelonephritis, and rupture of cysts into the retroperitoneal space may cause a fever that can last for weeks. Hepatic cysts may cause right upper quadrant pain if they enlarge or become infected.
Valvular disorders rarely cause symptoms but occasionally cause heart failure and require valvular replacement.
Symptoms and signs of unruptured cerebral aneurysm can be absent or may include headache, nausea and vomiting, and cranial nerve deficits; these manifestations warrant immediate intervention.
Signs are nonspecific and include hematuria and hypertension (each in about 40 to 50%) and, in 20% of patients, proteinuria in the subnephrotic range (< 3.5 g/24 hours in adults). Anemia is less common than in other types of chronic kidney disease, presumably because erythropoietin production is preserved. In advanced disease, the kidneys may become grossly enlarged and palpable, causing fullness in the upper abdomen and flank.
Diagnosis of ADPKD
Ultrasonography
Sometimes CT or MRI or genetic testing
The diagnosis of polycystic kidney disease is suspected in patients with the following:
A positive family history
Typical symptoms or signs
Cysts detected incidentally on imaging studies
Patients should be counseled before undergoing diagnostic testing, particularly if they are asymptomatic. For example, many authorities recommend against testing asymptomatic young patients because no disease-modifying treatment is effective at this age and diagnosis has potential negative effects on mood and on the ability to obtain life insurance on favorable terms.
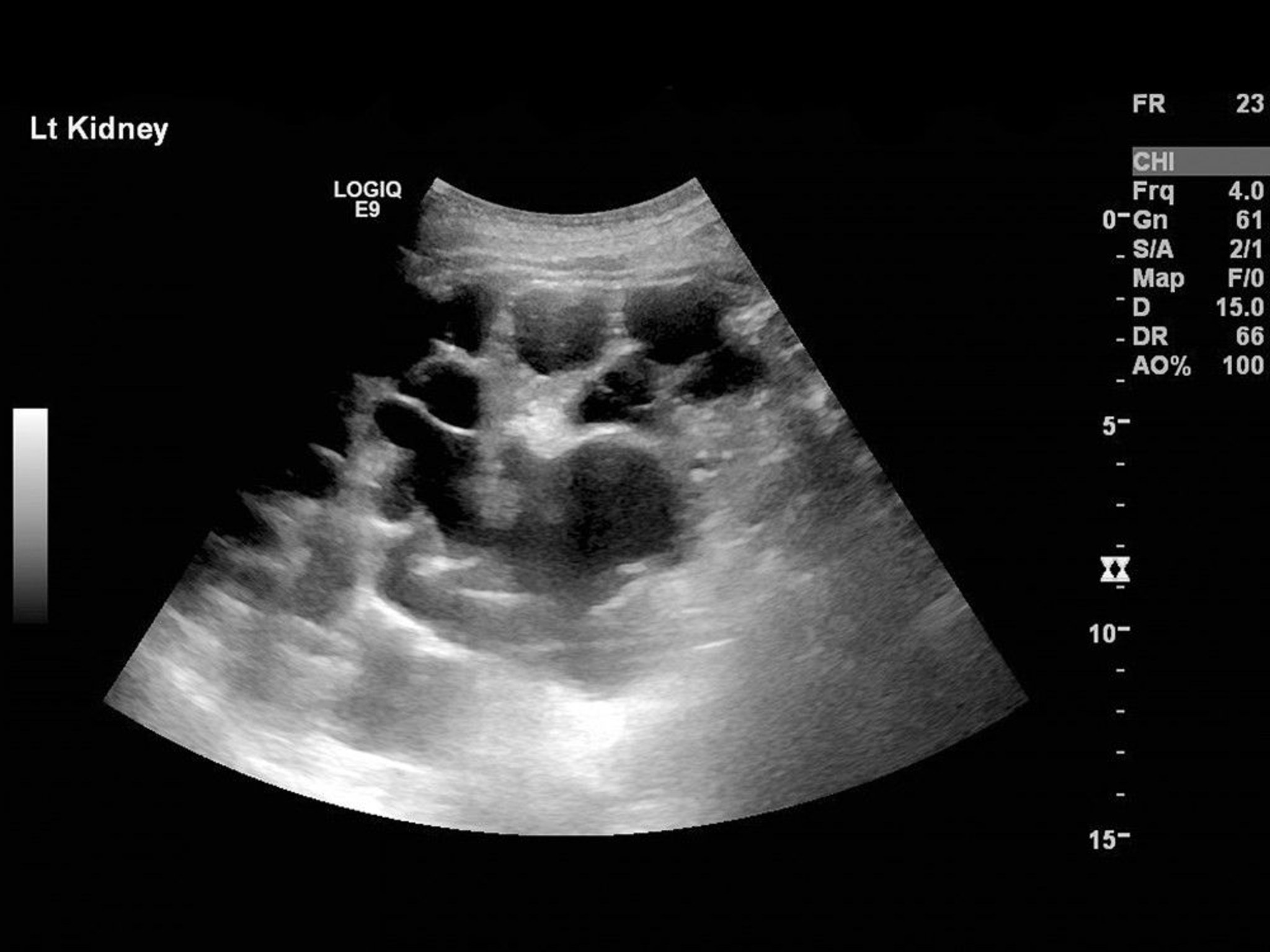
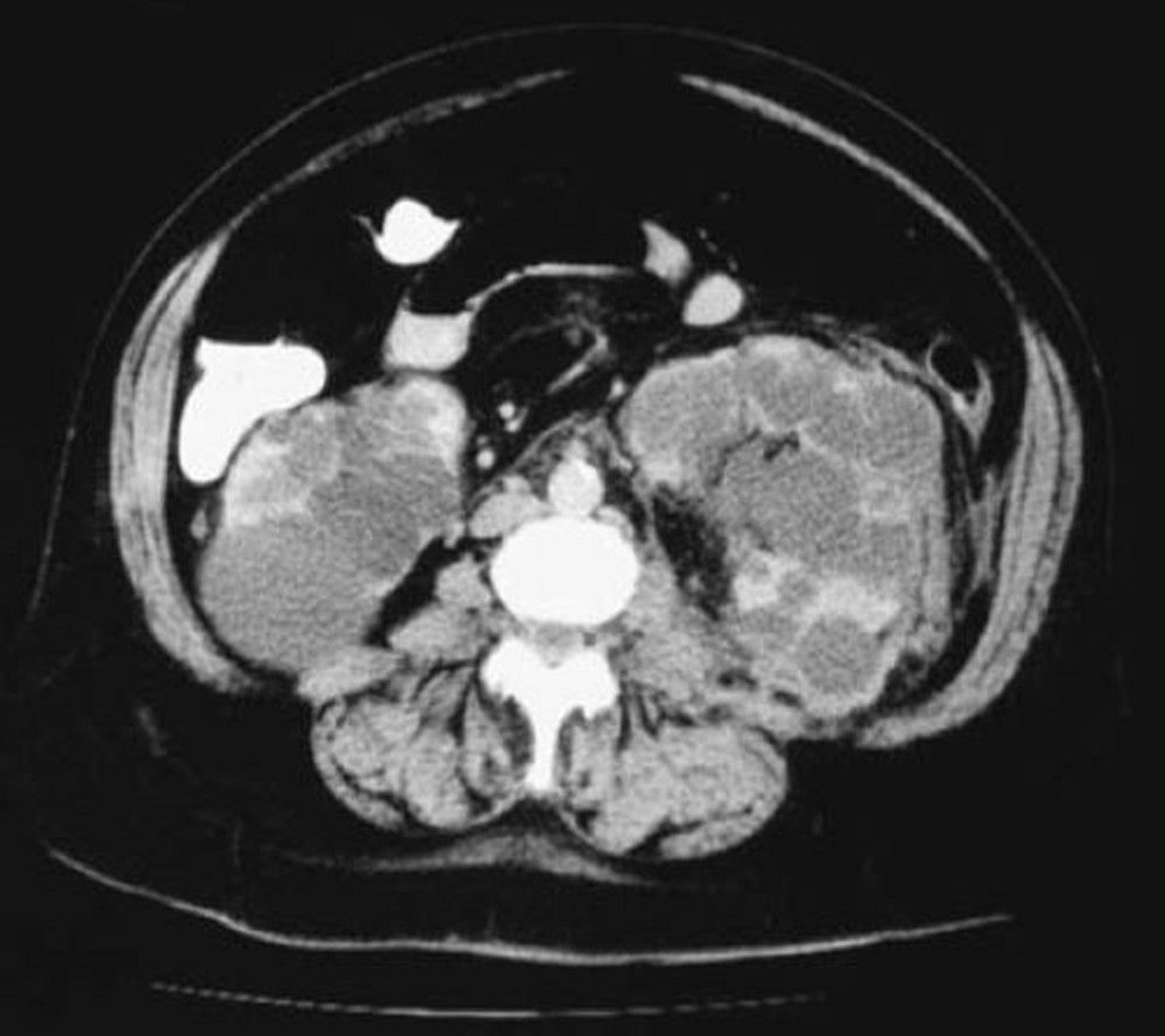
Diagnosis is usually by imaging, showing extensive and bilateral cystic changes throughout the kidneys, which are typically enlarged and have a moth-eaten appearance due to cysts that displace functional tissue. These changes develop with age and are less often present or obvious in younger patients.
Ultrasonography is usually done first. For patients with family history of ADPKD, ultrasound criteria (based on age and number of renal cysts) are used to diagnose or exclude ADPKD. Imaging criteria are not established in patients with negative or unknown history. CT or MRI are often done after establishing the diagnosis of ADPKD. CT and MRI are more sensitive than ultrasound in detecting cysts and can be useful in equivocal cases and for measuring cyst and kidney volume, which can have prognostic implications.
Urinalysis, renal function tests, and complete blood count (CBC) are done, but results are not specific.

SCIENCE PHOTO LIBRARY

© Springer Science+Business Media
Urinalysis detects mild proteinuria and microscopic or macroscopic hematuria. Gross hematuria may be due to a dislodged calculus or to hemorrhage from a ruptured cyst. Pyuria is common even without bacterial infection; thus diagnosis of infection should be based on culture results and clinical findings (eg, dysuria, fever, flank pain) as well as urinalysis. Initially, blood urea nitrogen (BUN) and creatinine are normal or only mildly elevated, but they slowly increase, especially when hypertension is present. Rarely, CBC detects polycythemia.
Patients with symptoms of cerebral aneurysm require high-resolution CT or magnetic resonance angiography. However, most experts do not recommend routine screening for cerebral aneurysm in asymptomatic patients. A reasonable approach is to screen patients with autosomal dominant polycystic kidney disease (ADPKD) who have a family history of hemorrhagic stroke or cerebral aneurysm.
Genetic testing for polycystic kidney disease (PKD) mutations is currently reserved for any of the following:
Patients with suspected PKD and no known family history
Patients with inconclusive results on imaging
Younger patients (eg, age < 30, in whom imaging results are often inconclusive) in whom the diagnosis must be made (eg, a potential kidney donor)
Genetic counseling is recommended for 1st-degree relatives of patients with ADPKD.
Treatment of ADPKD
Control of complications (eg, hypertension, infection, renal failure)
Supportive measures
Vasopressin antagonism or suppression
Strict control of hypertension is essential. Typically an angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker is used. In addition to controlling blood pressure, these medications help block angiotensin and aldosterone, growth factors that contribute to renal scarring and loss of renal function. Urinary tract infections (UTIs) should be treated promptly. Percutaneous aspiration of cysts may help relieve severe pain due to hemorrhage or compression but has no effect on long-term outcome. Nephrectomy is an option to relieve severe symptoms due to massive kidney enlargement (eg, pain, hematuria) or recurrent UTIs.
Hemodialysis, peritoneal dialysis, or kidney transplantation is required in patients who develop chronic renal failure. ADPKD does not recur in grafts.
Supportive measures include increased fluid intake (particularly water) to suppress vasopressin release, even if only partially, in patients who are able to safely excrete the load.
vasopressin receptor 2 antagonist, is a medication that may benefit patients with ADPKD (1, 2
3).
Treatment references
1. Torres VE, Chapman AB, Devuyst ON Engl J Med 367(25):2407-2418, 2012. doi: 10.1056/NEJMoa1205511
2. Torres VE, Chapman AB, Devuyst ON Engl J Med 16;377(20):1930-1942, 2017. doi: 10.1056/NEJMoa1710030
3. Cadnapaphornchai MA, George DM, McFann K, et alClin J Am Soc Nephrol 9(5):889-896, 2014.
Prognosis for ADPKD
Renal failure develops in 35 to 45% of patients with autosomal dominant polycystic kidney disease (ADPKD) by age 60. By age 75, 50 to 75% of patients require renal replacement therapy (dialysis or transplantation). On average, glomerular filtration rate (GFR) declines by about 5 mL/minute/year after the fourth decade of life. Predictors of more rapid progression to renal failure include the following:
Earlier age at diagnosis
Male sex
Sickle cell trait
PKD1 genotype
Larger or rapidly increasing kidney size
Gross hematuria
Black race
Increasing proteinuria
Cyst and kidney volume measurements predict risk of progression to chronic kidney disease>1500 mL (1).
Phosphaturic hormone fibroblast growth factor (FGF) 23 elevation was associated with increased kidney size and annualized rate of estimated glomerular filtration rate (eGFR) decline but interestingly did not enhance risk prediction for disease progression (2).
ADPKD does not increase risk of renal cancer, but if patients with ADPKD develop renal cancer, it is more likely to be bilateral. Renal cancer rarely causes death. Patients usually die of heart disease (sometimes valvular), disseminated infection, or ruptured cerebral aneurysm.
Prognosis references
1. Grantham JJ, Torres VE, Chapman AB, et al: Volume progression in polycystic kidney disease. N Engl J Med 18;354(20):2122-2130, 2006. doi: 10.1056/NEJMoa054341
2. Chonchol M, Gitomer B, Isakova T, et al: Fibroblast growth factor 23 and kidney disease progression in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 12(9):1461-1469, 2017. doi: 10.2215/CJN.12821216
Key Points
Autosomal dominant polycystic kidney disease occurs in about 1/1000 people.
About half of patients have no manifestations, but in others symptoms of back or abdominal pain, hematuria and/or hypertension develop gradually, usually beginning before age 30; 35 to 45% develop renal failure by age 60.
Extrarenal manifestations are common and include cerebral and coronary artery aneurysms, cardiac valve disease, and cysts in the liver, pancreas, and intestines.
Diagnose PKD based on imaging studies and clinical findings, reserving genetic testing for patients with no family history or with inconclusive results on imaging, or in young patients where the results may affect kidney donor eligibility. Expert opinion is advised prior to obtaining genetic testing due to various implications.
Do not routinely screen asymptomatic patients for ADPKD or asymptomatic patients who have ADPKD for cerebral aneurysms.
Arrange genetic counseling for 1st-degree relatives of patients with ADPKD.






