


(See also Overview of Platelet Disorders.)
Immune thrombocytopenia usually results from development of an autoantibody directed against a structural platelet antigen. These antiplatelet antibodies lead to increased platelet destruction, usually in the spleen, and inhibition of platelet production and release from megakaryocytes. In childhood ITP, the autoantibody may be triggered by viral antigens. The trigger in adults is unknown, although in some countries (eg, Japan, Italy), ITP has been associated with Helicobacter pylori infection, and treatment of the infection has been followed by remission of the ITP (1). COVID-19 infection rarely causes ITP, but COVID-19 vaccination may worsen thrombocytopenia in 2 to 12% of patients with ITP. ITP tends to worsen during pregnancy and increases the risk of maternal morbidity.
General reference
1. Kuter DJ: The treatment of immune thrombocytopenia (ITP)—focus on thrombopoietin receptor agonists. Annals of Blood Volume 6 March 2021
Symptoms and Signs of ITP
Although often asymptomatic and identified only by a low platelet count on a routine assay, when present the symptoms and signs of immune thrombocytopenia are
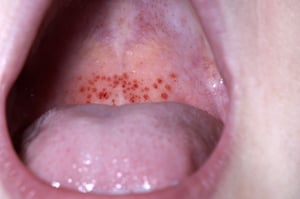
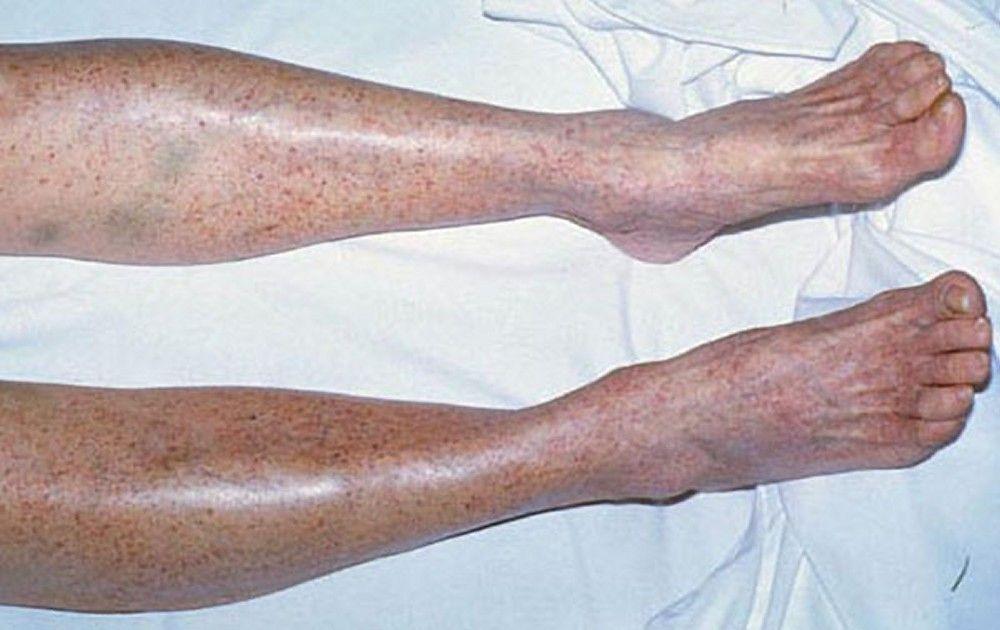
Petechiae
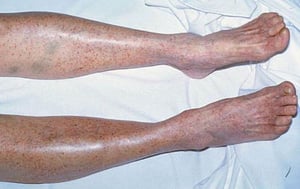
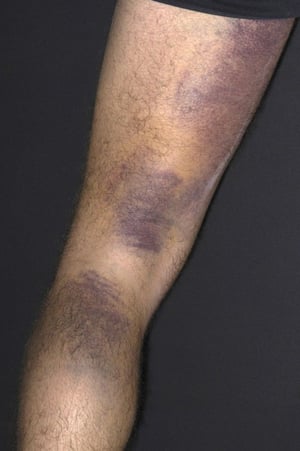
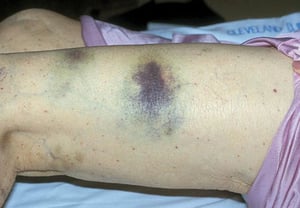
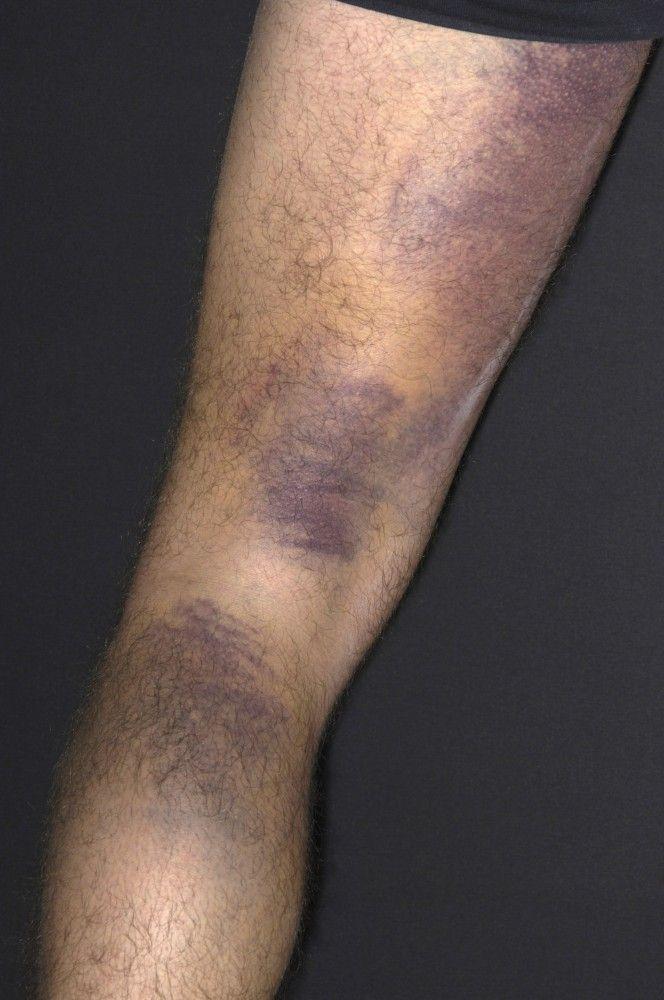
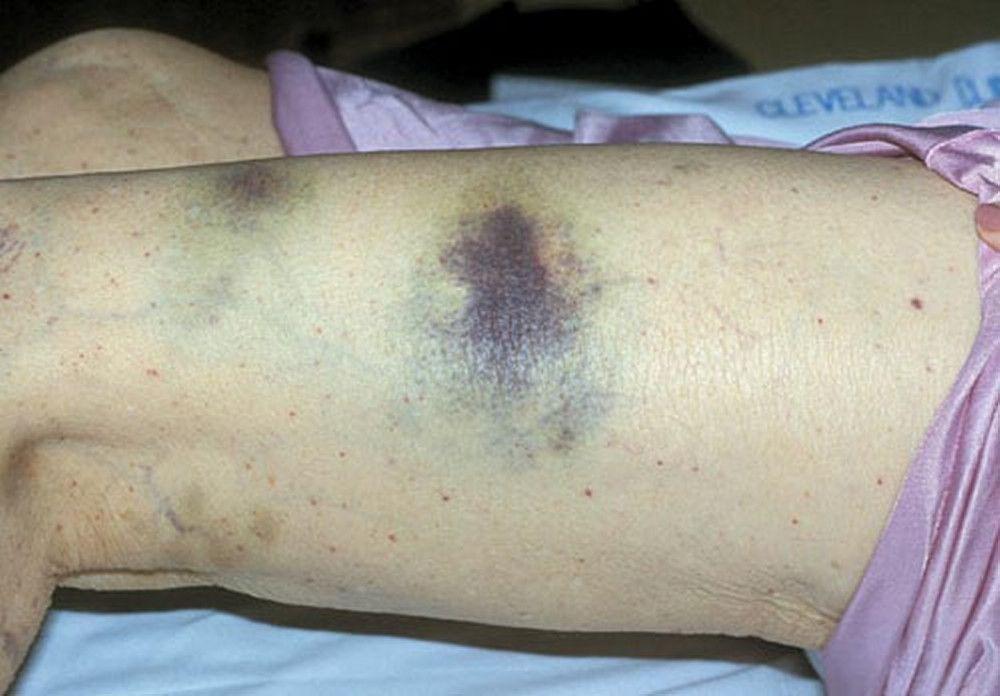
Purpura and/or ecchymoses
Mucosal bleeding
Increased menstrual bleeding
Gross gastrointestinal (GI) bleeding and hematuria are less common. The spleen is of normal size unless it is enlarged by a coexisting viral infection or autoimmune hemolytic anemia (Evans syndrome). Like the other disorders of increased platelet destruction, ITP is also associated with an increased risk of thrombosis.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

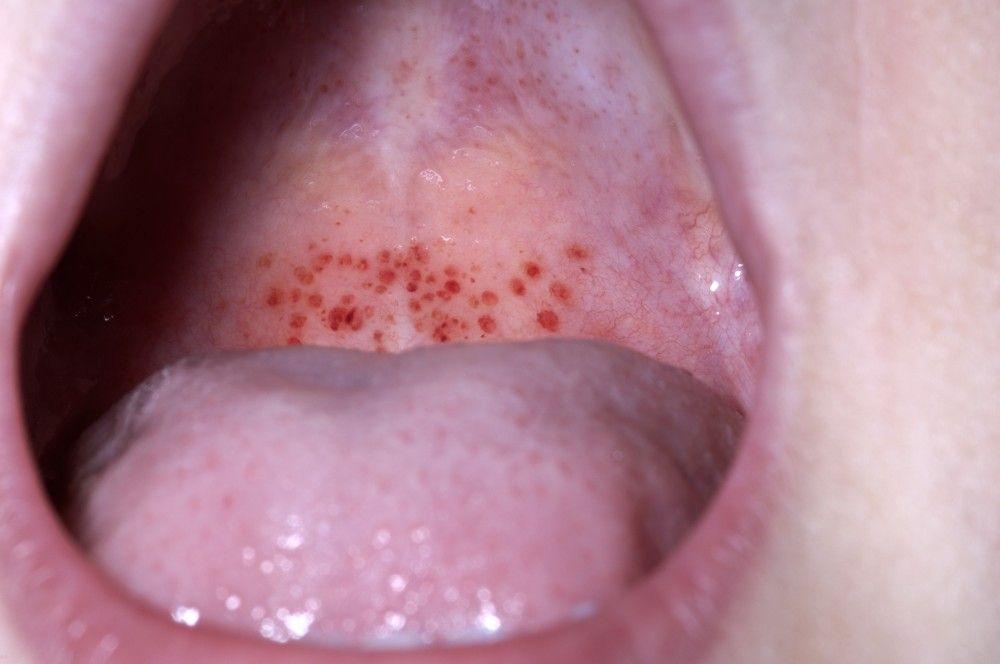
DR P. MARAZZI/SCIENCE PHOTO LIBRARY

By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

DR P. MARAZZI/SCIENCE PHOTO LIBRARY

By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
Diagnosis of ITP
Complete blood count (CBC) with platelets and peripheral blood smear
Rarely bone marrow aspiration
Exclusion of other thrombocytopenic disorders
Immune thrombocytopenia is suspected in patients with isolated thrombocytopenia (ie, otherwise normal CBC and peripheral blood smear). Because manifestations of immune thrombocytopenia (ITP) are nonspecific, other reversible causes of isolated thrombocytopenia (eg, drugs, alcohol, lymphoproliferative disorders, other autoimmune diseases, viral infections) need to be excluded by clinical evaluation and appropriate testing. Typically, patients have coagulation studies, liver tests, and tests for infection with hepatitis C and HIV. Peripheral blood smear must be reviewed to assess platelet size and granularity and to help exclude other major causes of thrombocytopenia such as thrombotic thrombocytopenic purpura (TTP), inherited thrombocytopenia, and leukemia. Testing for antiplatelet antibodies may aid the diagnosis in some cases (1). The immature platelet fraction may be elevated in ITP when the platelet count is < 50,000/mcL (< 50 × 109/L).
Bone marrow examination is not required to make the diagnosis but is done if blood counts or blood smear reveals abnormalities in addition to thrombocytopenia, when clinical features are not typical, or if patients fail to respond to standard therapies (eg, corticosteroids). In patients with ITP, bone marrow examination reveals normal or possibly increased numbers of megakaryocytes in an otherwise normal bone marrow sample.
Diagnosis reference
1. Al-Samkari H, Rosovsky RP, Karp Leaf RS: A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv 4(1):9–18, 2020. doi: 10.1182/bloodadvances.2019000868
Prognosis for ITP
Children typically recover spontaneously, even from severe thrombocytopenia, in several weeks to months.
In adults, spontaneous remission occurs in under 10%. With completion of initial treatment, about one-third of patients undergo remission. Up to 75% of patients improve within 5 years (1). However, many patients have mild and stable disease (ie, platelet counts > 30,000/mcL [> 30 × 109/L]) with minimal or no bleeding; they are often discovered by the automated platelet counting now routinely done with complete blood count.
Prognosis reference
1. Sailer T, Lechner K, Panzer S, et al: The course of severe autoimmune thrombocytopenia in patients not undergoing splenectomy. Haematologica 91:1041–1045, 2006.
Treatment of ITP
Oral corticosteroids
IV immune globulin (IVIG)
IV anti-D immune globulin
Sometimes, splenectomy
Thrombopoietin receptor agonists (TPO-RA)
Other immunosuppressants
For severe bleeding, IVIG, IV anti-D immune globulin, IV corticosteroids, and/or platelet transfusions
2019 guidelines are now available (1,2). Asymptomatic patients with a platelet count > 30,000/mcL (> 30 × 109/L) and no bleeding do not require treatment and can monitored.
Adults with newly diagnosed ITP with bleeding and a platelet count < 30,000/mcL (< 30 × 109
life-threatening bleeding in immune thrombocytopenia (ITP) but are rarely used for chronic treatment because their response may last for only a few days to weeks.
Splenectomy can achieve a complete remission in about two thirds of patients who relapse after initial corticosteroid therapy. Splenectomy is usually reserved for patients with severe thrombocytopenia (eg, < 15,000/mcL [< 15 × 109/L]) in whom bleeding risk cannot be controlled with medical therapy or those whose disease persists after 12 months. If thrombocytopenia can be controlled with second-line medical therapies, splenectomy is often not necessary (1, 2). Splenectomy results in an increased risk of thrombosis and infection (particularly with encapsulated bacteria such as pneumococcus); patients require vaccination against Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis (ideally > 2 weeks before the procedure).
Second-line medical therapies
Second-line medical therapies are available for patients with immune thrombocytopenia
Who are seeking to defer splenectomy in hope of a spontaneous remission
Who are not candidates for or refuse splenectomy
In whom splenectomy has not been effective
Such patients usually have platelet counts < 10,000 to 20,000/mcL (< 10 to 20 × 109
> 50,000/mcL (> 50 × 109/L), but data suggest that one third of adults will undergo a treatment-free remission after 1 year and > 50% after 2 years.
2 IV once a week for 4 weeks) has a response rate of 57%, but only 21% of adult patients remain in remission after 5 years (3
9/L—4).
Life-threatening bleeding in ITP
In children or adults with immune thrombocytopenia and life-threatening bleeding, rapid phagocytic blockade is attempted by giving IVIG 1 g/kg once a day for 1 to 2 days or, in Rh-positive patients, a single dose of IV anti-D immune globulin 75 mcg/kg. IV anti-D immunoglobulin is only effective in patients who have not had a splenectomy and may be associated with severe complications such as severe hemolysis and disseminated intravascular coagulation. This treatment usually causes the platelet count to rise within 2 to 4 days, but the count remains high for only 2 to 4 weeks.
2; maximum dose of 2 mg) has also been used in emergency situations but may produce neuropathy with repeated administration.
Early use of TPO-RA may also be effective in combination with the therapies above (5, 6).
Treatment of children with ITP
Treatment of children with immune thrombocytopenia is usually supportive because most children spontaneously recover. Even after months or years of thrombocytopenia, most children have spontaneous remissions. If mucosal bleeding occurs, corticosteroids or IVIG may be given. Corticosteroid and IVIG use is controversial because the increased platelet count may not improve clinical outcome. Splenectomy is rarely done in children. However, if thrombocytopenia is severe and symptomatic for >
Treatment references
1. Neunert C, Terrell DR, Arnold DM, et al: American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv 3(23):3829–3866, 2019. doi: 10.1182/bloodadvances.2019000966
2. Provan D, Arnold DM, Bussel JB, et al: Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv 3(22):3780–3817, 2019. doi: 10.1182/bloodadvances.2019000812
3. Patel VL, Mahevas M, Lee SY, et al: Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 119:5989–5995, 2012. doi: 10.1182/blood-2011-11-393975
4. Bussel J, Arnold DM, Grossbard E, et al: Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo‐controlled trials. Am J Hematol 93: 921–930, 2018. doi: 10.1002/ajh.25125
5. Kuter DJ, Tarantino MD, Lawrence TBlood Rev 49:100811, 2021. doi: 10.1016/j.blre.2021.100811
6. Lozano ML, Godeau B, Grainger J, et al: Romiplostim in adults with newly diagnosed or persistent immune thrombocytopenia. Expert Rev Hematol 13(12):1319–1332, 2020. doi: 10.1080/17474086.2020.1850253
Key Points
In immune thrombocytopenia (ITP), the immune system destroys platelets in the circulation and at the same time may attack bone marrow megakaryocytes, thereby reducing platelet production.
Other causes of isolated thrombocytopenia (eg, drugs, alcohol, lymphoproliferative disorders, other autoimmune diseases, viral infections) need to be excluded.
Children usually have spontaneous remission; in adults, spontaneous remission may occur during the first year but is less common (about 33%) than in children.
Corticosteroids (and sometimes intravenous immune globulin (IVIG) or IV anti-D immune globulin) are first-line treatments for bleeding or severe thrombocytopenia.
Bone marrow biopsy is typically not needed unless there are other concerning red or white blood cell abnormalities or in patients being considered for splenectomy who have not responded to standard treatment with corticosteroids or IVIG.
Thrombopoietin receptor agonists are highly effective in maintaining a safe platelet count in > 85% of adults.
COVID-19 infection rarely causes ITP, but COVID-19 vaccination may worsen thrombocytopenia in 2 to 12% of patients with ITP.
Splenectomy is often effective but is reserved for patients in whom medical therapy is ineffective or those whose disease persists after 12 months.
Platelet transfusion is given only for life-threatening bleeding.






