




La hemocromatosis hereditaria es un trastorno genético caracterizado por acumulación excesiva de hierro (Fe) que provoca daño tisular. Las manifestaciones pueden incluir síntomas sistémicos, trastornos hepáticos, miocardiopatía, diabetes, disfunción eréctil y artropatía. El diagnóstico se basa en el hallazgo de niveles elevados de ferritina sérica, hierro y saturación de transferrina y es confirmado con un análisis génico. Por lo general, el tratamiento consiste en flebotomías seriadas.
(Véase también Generalidades sobre la sobrecarga de hierro).
Etiología de la hemocromatosis hereditaria
Hay 4 tipos de hemocromatosis hereditaria, del 1 al 4, dependiendo del gen que está mutado.
Tipo 1: mutaciones del gen HFE (human homeostatic iron regulator)
Tipo 2 (hemocromatosis juvenil): mutaciones de los genes HJV (hemojuvelin BMP co-receptor) y HAMP (hepcidin antimicrobial peptide)
Tipo 3: mutaciones del gen TFR2 (transferrin receptor 2)
Tipo 4 (enfermedad de la ferroportina): mutaciones del gen SLC40A1 (solute carrier family 40 member 1)
Otros trastornos genéticos mucho más raros pueden causar sobrecarga hepática de hierro, pero el cuadro clínico generalmente está dominado por síntomas y signos debidos a la falla de otros órganos (p. ej., la anemia en la hipotransferrinemia o atransferrinemia o defectos neurológicos en la aceruloplasminemia).
Aunque estos tipos varían mucho en cuanto a edad de comienzo, las consecuencias clínicas de la sobrecarga de hierro son las mismas en todos los tipos (1).
Hemocromatosis hereditaria tipo 1
El tipo 1 es la hemocromatosis hereditaria clásica, también llamada hemocromatosis relacionada con el gen HFE. Más del 80% de los casos son causados por la mutación homocigota C282Y o por la mutación heterocigota compuesta C282Y/H63D. Las mutaciones homocigotas H63D ocurren rara vez y tienen el mismo fenotipo que los casos homocigotos C282y. El trastorno es autosómico recesivo, con una frecuencia homocigota de 1:200 y una frecuencia heterocigota de 1:8 en personas con ascendencia europea septentrional. Las mutaciones C282Y y H63D son infrecuentes en personas con ascendencia africana y raras en personas con ascendencia asiática. De los pacientes con características clínicas de hemocromatosis, el 83% son homocigotos. Sin embargo, por razones desconocidas, la enfermedad fenotípica (clínica) es mucho menos frecuente que la prevista por la frecuencia del gen (es decir, numerosos individuos homocigotos no manifiestan el trastorno).
Hemocromatosis hereditaria tipo 2
La hemocromatosis hereditaria tipo 2 (hemocromatosis juvenil) es un trastorno autosómico recesivo raro causado por mutaciones del gen HJV que afectan la transcripción de la proteína hemojuvelina o mutaciones en el gen HAMP, que codifica directamente la hepcidina. A menudo aparece en adolescentes.
Hemocromatosis hereditaria tipo 3
Las mutaciones del gen que codifica el receptor de transferrina 2 (TFR2), una proteína que parece controlar la saturación de transferrina, puede causar una forma autosómica recesiva rara de hemocromatosis.
Hemocromatosis hereditaria tipo 4
La hemocromatosis hereditaria tipo 4 (enfermedad de la ferroportina) afecta, en gran medida, a personas con ascendencia europea meridional. Se debe a una mutación autosómica dominante del gen SLC40A1 y afecta la capacidad de la ferroportina para unirse a la hepcidina.
Deficiencia de transferrina y ceruloplasmina
En la deficiencia de transferrina (hipotransferrinemia o atransferrinemia), el hierro absorbido que ingresa en el sistema porta no unido a transferrina se deposita en el hígado. Hay reducción del traslado ulterior de hierro a los sitios de producción de eritrocitos debido a la deficiencia de transferrina.
En la deficiencia de ceruloplasmina (aceruloplasminemia), la falta de ferroxidasa causa conversión defectuosa de Fe2+ a Fe3+; esta conversión es necesaria para la unión a transferrina. La defectuosa unión a transferrina altera el desplazamiento de hierro de los depósitos intracelulares al transporte plasmático, con la consiguiente acumulación de hierro en los tejidos.
Referencia de la etiología
1. Pietrangelo A: Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 139:393–408, 2010.
Fisiopatología de la hemocromatosis hereditaria
El contenido corporal total normal de hierro es de alrededor de 2,5 g en las mujeres y de 3,5 g en los hombres. Como los síntomas pueden no aparecer hasta que la acumulación de hierro es excesiva (p. ej., > 10 a 20 g), a veces no se reconoce la hemocromatosis hasta más tarde en la vida, aun cuando es una anomalía hereditaria. En las mujeres, las manifestaciones clínicas son infrecuentes antes de la menopausia, porque la pérdida de hierro por la menstruación (y, en ocasiones, por el embarazo y el parto) tiende a compensar su acumulación.
El mecanismo de sobrecarga de hierro tanto en la hemocromatosis HFE como en la no HFE consiste en aumento de su absorción digestiva, que causa depósito crónico de hierro en los tejidos. La hepcidina, un péptido derivado del hígado, es el mecanismo de control crucial para la absorción de hierro. La hepcidina normalmente expeimenta una regulación positiva cuando las reservas de hierro son elevadas y, a través de su efecto inhibidor sobre la ferroportina (que participa en la absorción de hierro), impide la absorción excesiva de hierro y su almacenamiento en las personas normales. La hemocromatosis tipos 1 a 4 comparten la misma base patogénica (p. ej., la falta de síntesis o de actividad de hepcidina) y características clínicas clave.
Por lo general, la lesión tisular parece deberse a radicales libres hidroxilo reactivos, generados cuando el depósito de hierro en los tejidos cataliza su formación. Otros mecanismos pueden afectar órganos particulares (p. ej., la hiperpigmentación cutánea puede deberse a aumento de melanina, así como a acumulación de hierro). En el hígado, la peroxidación lipídica asociada al hierro induce la apoptosis de los hepatocitos, que estimula la activación de las células de Kupffer y la liberación de citocinas proinflamatorias. Estas citocinas activan a las células estrelladas hepáticas para producir colágeno, lo que resulta en una acumulación patológica de fibrosis hepática y riesgo de carcinoma hepatocelular.
Signos y síntomas de la hemocromatosis hereditaria
Las consecuencias clínicas de sobrecarga de hierro tienden a ser similares, independientemente de la etiología y la fisiopatología de la sobrecarga.
Tradicionalmente, los especialistas consideraban que no aparecían síntomas hasta que se habían producido lesiones orgánicas. Sin embargo, el daño de los órganos es lento y sutil, y suele haber síntomas y signos inespecíficos y cansancio en estadios tempranos. Por ejemplo, la disfunción hepática puede manifestarse de manera insidiosa con fatiga, dolor abdominal en el cuadrante superior derecho y hepatomegalia. Las alteraciones de las pruebas de laboratorio en la sobrecarga de hierro y la hepatitis generalmente preceden a los síntomas.
En la hemocromatosis hereditariatipo 1 (HFE) los síntomas se relacionan con los órganos que presentan los depósitos de hierro más grandes (véase tabla Manifestaciones frecuentes de la hemocromatosis hereditaria). En los hombres, los síntomas iniciales pueden ser hipogonadismo y disfunción eréctil causados por depósito gonadal. La intolerancia a la glucosa o la diabetes mellitus es otra presentación inicial común. Algunos pacientes presentan hipotiroidismo.
La hepatopatía es la complicación más frecuente y puede evolucionar a cirrosis; del 20 al 30% de los pacientes con cirrosis presentan carcinoma hepatocelular. La cirrosis hepática es la causa más frecuente de muerte. El aumento de las enzimas hepáticas es una de las anomalías de laboratorio más comunes en pacientes con hemocromatosis tipo 1.
La miocardiopatía con insuficiencia cardíaca es la segunda complicación fatal en orden de frecuencia. La hiperpigmentación (diabetes bronceada) y la porfiria cutánea tardía son comunes, al igual que la artropatía sintomática en las manos.
En la enfermedad tipo 2, los signos y síntomas son hepatomegalia progresiva e hipogonadismo hipogonadotrópico.
En la enfermedad tipo 3 los síntomas y signos son similares a los de la hemocromatosis hereditaria tipo 1 (HFE).

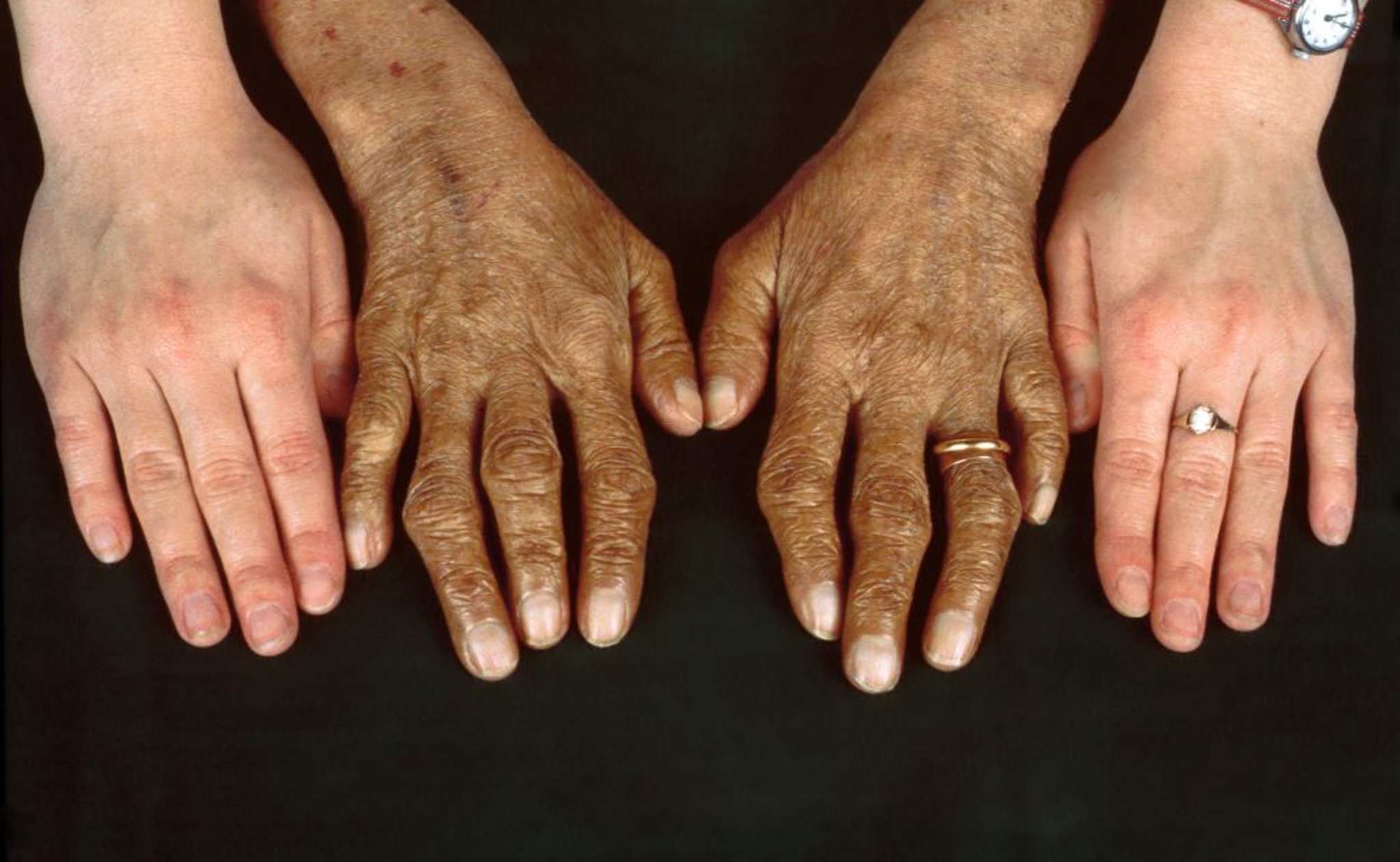
Clinical Photography/SCIENCE PHOTO LIBRARY
La enfermedad tipo 4 se manifiesta en la primera década de la vida por aumento de las concentraciones de ferritina, con saturación de transferrina normal o baja; cuando los pacientes cursan la tercera y cuarta décadas de la vida se observa una saturación progresiva de la transferrina. Las manifestaciones clínicas son más leves que en la enfermedad tipo 1, con hepatopatía modesta y anemia leve.
Diagnóstico de la hemocromatosis hereditaria
Concentración sérica de ferritina, hierro sérico en ayunas y saturación de transferrina
Estudios genéticos
En ocasiones, biopsia hepática
Los signos y síntomas pueden ser inespecíficos, sutiles y de comienzo gradual, de manera que el médico debe tener en cuenta la posibilidad de este diagnóstico. Debe sospecharse hemocromatosis primaria cuando las manifestaciones típicas, en particular combinaciones de ellas, continúan siendo inexplicables después de la evaluación de rutina. Los antecedentes familiares de hemocromatosis, cirrosis o carcinoma hepatocelular son una pista más específica. En todos los pacientes con hepatopatía crónica se debe evaluar la posibilidad de sobrecarga de hierro, independientemente de su raza o etnia
La determinación de ferritina sérica es la prueba inicial más simple y más directa. En la hemocromatosis hereditaria, suele haber altas concentraciones (> 200 ng/mL [> 200 mcg/L] en mujeres o > 250 ng/mL [> 250 mcg/L] en hombres), pero estas pueden deberse a otras alteraciones, como trastornos inflamatorios hepáticos (p. ej., hepatitis viral crónica, esteatohepatitis no alcohólica, hepatopatía alcohólica), cáncer, ciertos trastornos inflamatorios sistémicos (p. ej., artritis reumatoide, linfohistiocitosis hemofagocítica) u obesidad. Si la concentración de ferritina es anormal, se realizan más estudios, como hierro sérico en ayunas (en general, > 300 mg/dL [> 53,7 mcmol/L]) y capacidad de fijación de hierro (saturación de transferrina; en general, niveles > 50%). Una saturación de transferrina < 45% tiene un valor predictivo negativo del 97% para la sobrecarga de hierro.
En la enfermedad tipo 2, las concentraciones de ferritina son > 1.000 ng/mL (> 1000 mcg/L), y la saturación de transferrina es > 90%.
En la deficiencia de transferrina o ceruloplasmina, las concentraciones séricas de transferrina (es decir, capacidad de fijación del hierro) y ceruloplasmina están muy disminuidos o son indetectables.
El análisis genético confirma la hemacromatosis hereditaria causada por mutaciones del gen HFE. Aproximadamente el 70% de los pacientes con mutaciones homocigotas C282Y del gen HFE tiene un nivel elevado de ferritina, pero solo alrededor del 10% de estos pacientes tienen evidencia de disfunción orgánica. La sobrecarga de hierro clínicamente significativa es aún menos común en pacientes con mutaciones heterocigotas del gen HFE (es decir, C282Y/H63D). La hemocromatosis tipos 2 a 4 se sospecha en casos menos frecuentes en los que las pruebas de hierro y ferritina en sangre indican sobrecarga de hierro pero el estudio genético es negativo para la mutación del gen HFE, particularmente en pacientes más jóvenes. La confirmación de estos diagnósticos mediante pruebas genéticas no está disponible en forma sistemática.
Cuando se confirma el diagnóstico, el hígado debe ser evaluado para fibrosis y cirrosis. Hasta el 80% de los pacientes con cirrosis y una mutación homocigota en C282Y tendrán una ferritina > 1000 ng/mL, niveles elevados de AST (aspartato transaminasa) y ALT (alanina transaminasa) y un recuento de plaquetas < 200 x 103/mcL (< 200 × 109/L). Como la presencia de cirrosis incide en el pronóstico, cuando la ferritina es > 1.000 ng/mL, suele realizarse una biopsia hepática para determinar el contenido de hierro (cuando es factible). La biopsia hepática también se recomienda en pacientes con evidencia serológica de sobrecarga de hierro pero con una prueba genética negativa. La RM con elastografía por RM con contraste (ERM) es una alternativa no invasiva para estimar el contenido de hierro hepático que está volviéndose cada vez más precisa.
Se deben estudiar los familiares directos de pacientes con hemocromatosis hereditaria mediante determinación de las concentraciones séricas de ferritina e investigación de las mutaciones en el gen C282YH63D y HFE.
Tratamiento de la hemocromatosis hereditaria
Flebotomía
Está indicado el tratamiento en pacientes con manifestaciones clínicas, altas concentraciones séricas de ferritina (en particular, concentraciones > 1.000 ng/mL [> 1000 mcg/L]), o alta saturación de transferrina. Los pacientes asintomáticos solo requieren evaluación clínica periódica (p. ej., anual) y determinación de hierro y ferrritina séricos, saturación de transferrina y enzimas hepáticas.
La flebotomía es el método más simple y más eficaz para eliminar el exceso de hierro. Retrasa la progresión de fibrosis a cirrosis, y a veces, incluso revierte los cambios cirróticos y prolonga la supervivencia, pero no previene el carcinoma hepatocelular. Se extraen alrededor de 500 mL de sangre (aproximadamente, 250 mg de hierro) en forma semanal o bisemanal (semana por medio), hasta que las concentraciones séricas de ferritina alcanzan entre 50 y 100 ng/mL. Puede requerirse una flebotomía semanal o bisemanal durante muchos meses (p. ej., si se eliminan 250 mg de hierro por semana, se necesitarán 40 semanas para eliminar 10 g de hierro). Cuando las concentraciones de hierro son normales, las flebotomías pueden ser intermitentes para mantener una ferritina entre 50 y 100 ng/mL.
La diabetes mellitus, la miocardiopatía, la disfunción eréctil y otras manifestaciones secundarias se tratan según esté indicado. Pacientes con fibrosis o cirrosis avanzadas debido a sobrecarga de hierro deben ser examinados para descartar carcinoma hepatocelular cada 6 meses con ecografía hepática.
Los pacientes deben cumplir una dieta balanceada; no es necesario limitar el consumo de alimentos con hierro (p. ej., carne roja, hígado). Debe consumirse alcohol sólo con moderación, porque puede aumentar la absorción de hierro y, en altas cantidades, incrementa el riesgo de cirrosis. Deben evitarse los suplementos de vitamina C porque aumentan la absorción de hierro en el duodeno.
En los pacientes con la enfermedad tipo 4, hay mala tolerancia a la flebotomía; se requiere control seriado de la hemoglobina y la saturación de transferrina.
El tratamiento de la deficiencia de transferrina y la deficiencia de ceruloplasmina es experimental; p. ej., los quelantes del hierro pueden ser mejor tolerados que la flebotomía, porque los pacientes suelen presentar anemia.
Conceptos clave
Hay 4 tipos de hemocromatosis hereditaria, todos los cuales implican mutaciones que alteran la capacidad del cuerpo para inhibir la absorción de hierro cuando las reservas son excesivas.
Los efectos de la sobrecarga de hierro son similares en todos los tipos e incluyen enfermedad hepática (que conduce a la cirrosis), pigmentación de la piel, diabetes, artropatía, hipogonadismo y a veces insuficiencia cardíaca.
Diagnosticar mediante la medición del nivel de ferritina sérica; si está elevada, confirmar con la demostración de hierro sérico elevado, saturación de transferrina y evaluación genética.
Una vez hecho el diagnóstico, debe evaluarse el grado de fibrosis hepática mediante biopsia hepática o estudios de diagnóstico por imágenes no invasivos, como RM con elastografía por RM, para determinar el pronóstico; se debe considerar la indicación de pruebas genéticas y el cribado de los familiares de primer grado.
Tratar con flebotomía y moderación del consumo de alcohol.









