


La ataxia-telangiectasia se debe a un defecto de reparación del DNA que suele provocar inmunodeficiencia humoral y celular; causa ataxia cerebelosa progresiva, telangiectasias oculocutáneas e infecciones sinopulmonares recidivantes. Se miden IgA y concentración sérica de alfa-fetoproteína 1. Los estudios genéticos son necesarias para confirmar el diagnóstico. El tratamiento se realiza con antibióticos profilácticos o con inmunoglobulina.
(Véase también Generalidades sobre las inmunodeficiencias y Abordaje del paciente con un trastorno de inmunodeficiencia).
La ataxia-telangiectasia es trastorno de inmunodeficiencia primaria autosómico recesivo que implica deficiencias combinadas en las inmunidades humoral y celular. La incidencia estimada es de 1 cada 20.000 a 100.000 nacimientos. La ataxia-telangiectasia se debe a mutaciones del gen que codifica la proteína mutada de la ataxia-telangiectasia (ATM). ATM está implicado en la detección de daños en el DNA y ayuda a controlar la tasa de crecimiento y la división celular.
A menudo, los pacientes carecen de IgA e IgE y tienen un defecto progresivo de los linfocitos T.
Síntomas y signos de la ataxia-telangiectasia
El inicio de los síntomas neurológicos y los signos de inmunodeficiencia varían.
La ataxia suele ser el primer síntoma y en general se manifiesta cuando los niños comienzan a caminar. La progresión de los síntomas neurológicos conducen a una incapacidad grave. Aparecen balbuceo, movimientos coreoatetoides y nistagmo, y una debilidad muscular que suele progresar a atrofia.
Las telangiectasias pueden no aparecer hasta los 4 a 6 años de edad; son más prominentes en las conjuntivas bulbares, los pabellones auriculares, las fosas antecubitales y poplítea y los lados del cuello.
Las infecciones sinopulmonares recidivantes producen neumonías, bronquiectasias y una enfermedad pulmonar restrictiva crónica.
Pueden aparecer anomalías endocrinas (p. ej., la disgenesia gonadal, la atrofia testicular y la diabetes mellitus).
La frecuencia de cáncer (especialmente leucemia, linfoma, tumores cerebrales, y el cáncer gástrico) es alta. El cáncer se produce normalmente después de los 10 años y a una tasa de alrededor de 1%/año, pero es un riesgo para toda la vida y puede ocurrir a cualquier edad.

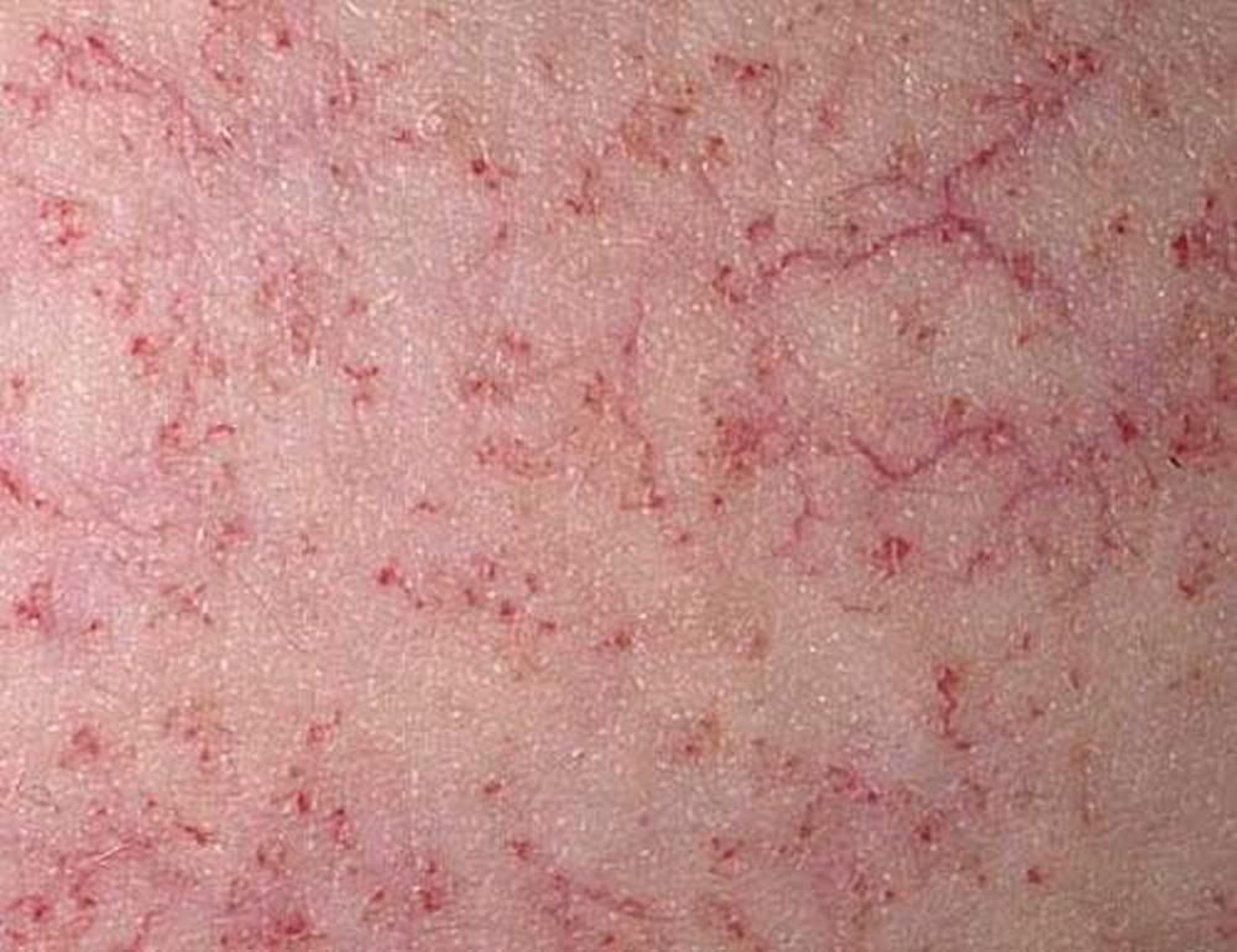
Imagen cortesía de Thomas Habif, MD.
Diagnóstico de la ataxia-telangiectasia
IgA y concentraciones sérica de alfa-fetoproteína 1
Estudios genéticos
Los siguientes hallazgos clínicos sugieren el diagnóstico de ataxia-telangiectasia:
Ataxia cerebelosa (en particular en presencia de telangiectasias)
Bajos niveles de IgA (presentes en el 80% de los pacientes con este trastorno)
Niveles séricos altos de alfa-fetoproteína 1
Si se realiza el análisis del cariotipo, a menudo se detectan roturas cromosómicas, compatibles con un defecto en la reparación del DNA.
El diagnóstico de la ataxia-telangiectasia se confirma mediante la identificación de mutaciones en ambos alelos del gen de la proteína ATM. Debido a que los portadores de una mutación ataxia-telangiectasia suelen permanecer asintomáticos, la evaluación de los hermanos para detectar un estado de portador puede ayudar a predecir sus posibilidades de tener un hijo afectado.
Las pruebas para detectar anormalidades endocrinológicas y cáncer se realizan sobre la base de la presentación clínica.
Se debe proponer asesoramiento genético a los familiares de los pacientes diagnosticados, con la opción de investigar el estado de portador de ataxia-telangiectasia, porque los portadores pueden tener un mayor riesgo de cáncer (1)
Referencia del diagnóstico
1. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, et al: Ataxia telangiectasia: a review. Orphanet J Rare Dis 11(1):159, 2016. doi:10.1186/s13023-016-0543-7
Tratamiento de la ataxia-telangiectasia
Tratamiento paliativo mediante el uso de antibióticos profilácticos o terapia de reposición de inmunoglobulina (Ig)
El tratamiento con antibióticos profilácticos o inmunoglobulina puede ser útil en los pacientes con ataxia-telagiectasia.
En un pequeño estudio, el tratamiento con amantadina resultó en una mejoría mínima en la función motora, pero no existe un tratamiento eficaz para el deterioro neurológico progresivo, que causa la muerte, generalmente hacia los 30 años (1).
La quimioterapia a menudo se indica para el tratamiento de cánceres asociados.
Referencia del tratamiento
1. Nissenkorn A, Hassin-Baer S, Lerman SF, et al: Movement disorder in ataxia-telangiectasia: treatment with amantadine sulfate. J Child Neurol 28(2):155–160, 2013. doi:10.1177/0883073812441999
Conceptos clave
La ataxia-telangiectasia incluye deficiencias combinadas en las inmunidades humoral y celular.
La ataxia suele ser el primer síntoma y en general se manifiesta cuando los niños comienzan a caminar.
No existe un tratamiento eficaz para el deterioro neurológico progresivo, que causa la muerte, por lo general a los 30 años.









