







Une glomérulonéphrite rapidement progressive est un syndrome néphritique aigu généralement accompagnée d'une formation visible en microscopie optique de croissants glomérulaires avec évolution vers une insuffisance rénale dans les semaines ou les mois qui suivent. Le diagnostic repose sur l'anamnèse, les analyses d'urine, les tests sérologiques et la biopsie rénale. Le traitement repose sur les corticostéroïdes, associés ou non au cyclophosphamide ou rituximab et parfois les échanges plasmatiques.
La glomérulonéphrite rapidement progressive, un type de syndrome néphritique, est un diagnostic anatomopathologique avec présence de formations glomérulaires étendues en croissant (c'est-à-dire que > 50% des glomérules du prélèvement contiennent des croissants); non traitée, la glomérulonéphrite rapidement progressive évolue en quelques semaines ou quelques mois jusqu'au stade de néphropathie terminale. Elle est relativement rare, touchant 10 à 15% des patients atteints de glomérulonéphrite et se manifeste préférentiellement chez le patient de 20 à 50 ans. Les types et causes sont classés par les signes en microscopie à immunofluorescence et par les tests sérologiques (p. ex., maladie à anticorps anti-membrane basale, anticorps anticytoplasme de neutrophiles [ANCA], voir tableau Classification sérologique de la glomérulonéphrite rapidement progressive d'après la microscopie en immunofluorescence).
Maladie des anticorps anti-membrane basale glomérulaire
La maladie des anticorps anti-membrane basale glomérulaire est une glomérulonéphrite auto-immune qui représente jusqu'à 10% des cas de glomérulonéphrite rapidement progressive. Elle peut résulter d'expositions respiratoires (p. ex., fumée de cigarette, infection virale des voies respiratoires supérieures) ou d'exposition à d'autres stimuli qui exposent le collagène capillaire alvéolaire aux agents toxiques en induisant ainsi la formation d'anticorps anticollagènes. Les anticorps anticollagènes interagissent avec la membrane basale glomérulaire en fixant le complément et en déclenchant généralement une réponse inflammatoire médiée par des cellules rénales et pulmonaires.
Le terme syndrome de Goodpasture correspond à l'association d'une glomérulonéphrite et d'une hémorragie alvéolaire en présence d'anticorps anti-membrane basale glomérulaire. La glomérulonéphrite sans hémorragie alvéolaire en présence d'anticorps anti-membrane basale glomérulaire est appelée glomérulonéphrite à anti-membrane basale glomérulaire. L'analyse en immunofluorescence du prélèvement de tissu biopsié montre des dépôts linéaires d'IgG.
Glomérulonéphrite rapidement progressive à complexes immuns
La glomérulonéphrite rapidement progressive à complexes immuns complique de nombreuses maladies infectieuses et rhumatologiques systémiques et se produit également avec d'autres glomérulopathies primaires.
L'immunofluorescence révèle des dépôts immuns granuleux non spécifiques. Cette maladie représente jusqu'à 40% des cas de glomérulonéphrite rapidement progressive. La physiopathologie reste habituellement inconnue.
Glomérulonéphrite rapidement progressive pauci-immune
La glomérulonéphrite rapidement progressive pauci-immunitaire se différencie par l'absence de complexes immuns ou de dépôts de complément en immunofluorescence. Elle constitue jusqu'à 50% des cas de glomérulonéphrite rapidement progressive. Presque tous les patients ont un taux élevé d'anticorps anti-cytoplasme de neutrophiles (ANCAs), généralement ANCA anti-protéinase-3 ou ANCA anti-myéloperoxydase et une vascularite systémique.
Maladie à double anticorps
La maladie à double anticorps est observée en présence d'anticorps anti-GBM et d'ANCA. Elle est rare.
Glomérulonéphrite rapidement progressive idiopathique
Les cas idiopathiques sont rares. Ils concernent les patients qui présentent l'un des signes suivants:
Complexes immunitaires mais aucune cause évidente telle qu'une infection, une maladie rhumatismale systémique ou un trouble glomérulaire
Signes pauci-immunitaires mais absence d'anticorps ANCA
Symptomatologie de la glomérulonéphrite rapidement progressive
Les symptômes sont habituellement insidieux, associant asthénie, fatigue, fièvre, nausées et vomissements, anorexie, arthralgie et douleurs abdominales. Certains patients présentent une symptomatologie identique à celle de la glomérulonéphrite post-infectieuse, avec hématurie soudaine. Environ 50% des patients présentent des œdèmes et on retrouve un épisode aigu de type grippal dans les 4 semaines ayant précédé l'installation de l'insuffisance rénale, habituellement suivie d'une oligurie sévère. Un syndrome néphrotique est présent dans 10 à 30% des cas. L'HTA est rare et rarement grave. Le patient atteint d'une maladie des anticorps anti-membrane basale glomérulaire peut développer une hémorragie pulmonaire, qui peut se manifester par une hémoptysie ou n'être détectable que par des signes évocateurs d'infiltrats alvéolaires diffus à l'imagerie pulmonaire (syndrome rénopulmonaire ou hémorragique alvéolaire diffus).
Diagnostic de la glomérulonéphrite rapidement progressive
Insuffisance rénale progressive sur des semaines ou mois
Sédiment urinaire de type néphritique
Tests sérologiques
Taux de complément sérique
Biopsie rénale
Le diagnostic est suspecté devant une lésion rénale aiguë en cas d'hématurie et de globules rouges dysmorphiques ou de cylindres de globules rouges. Le test comprend une créatininémie, des analyses d'urine, une NFS, des tests sérologiques et une biopsie rénale. Le diagnostic est établi habituellement au moyen de tests sérologiques et d'une biopsie rénale.
La créatininémie est presque toujours élevée.
Une analyse d'urine montre une hématurie toujours présente, et des cylindres de globules rouges sont habituellement présents. Un sédiment télescopé (c'est-à-dire, un sédiment avec de multiples éléments, dont des globules blancs, des globules rouges dysmorphiques et de larges cylindres granulaires et cireux de globules blancs et rouges) est fréquent.
Sur la NFS, l'anémie est habituellement présente et l'hyperleucocytose est fréquente.
Les tests sérologiques doivent comprendre la recherche des anticorps anti-membrane basale glomérulaire (maladie des anticorps anti-membrane basale glomérulaire); les anticorps antistreptolysine O (ASLO), les anticorps anti-ADN, ou les cryoglobulines (glomérulonéphrite rapidement progressive à complexe immuns); et les titres d'ANCA (antineutrophil cytoplasmic antibodies) (glomérulonéphrite rapidement progressive pauci-immunitaire).
Le dosage du complément (C3 et C4 sérique) peut être utile lorsqu'une glomérulonéphrite rapidement progressive à complexes immuns est suspectée, car l'hypocomplémentémie est fréquente.
Une biopsie rénale précoce est essentielle. La caractéristique commune à tous les types de glomérulonéphrite rapidement progressive est la prolifération focale de cellules épithéliales glomérulaires, mêlées dans certains cas à nombre de neutrophiles, formant un amas cellulaire (en forme de croissant) qui remplit la capsule de Bowman de > 50% des glomérules. Le flocculus glomérulaire apparaît habituellement hypocellulaire et collabé. Une nécrose du flocculus ou de la capsule de Bowman peut se produire et constituer l'anomalie la plus marquante. Chez ces patients, des éléments histologiques en faveur d'une vascularite doivent être recherchés.

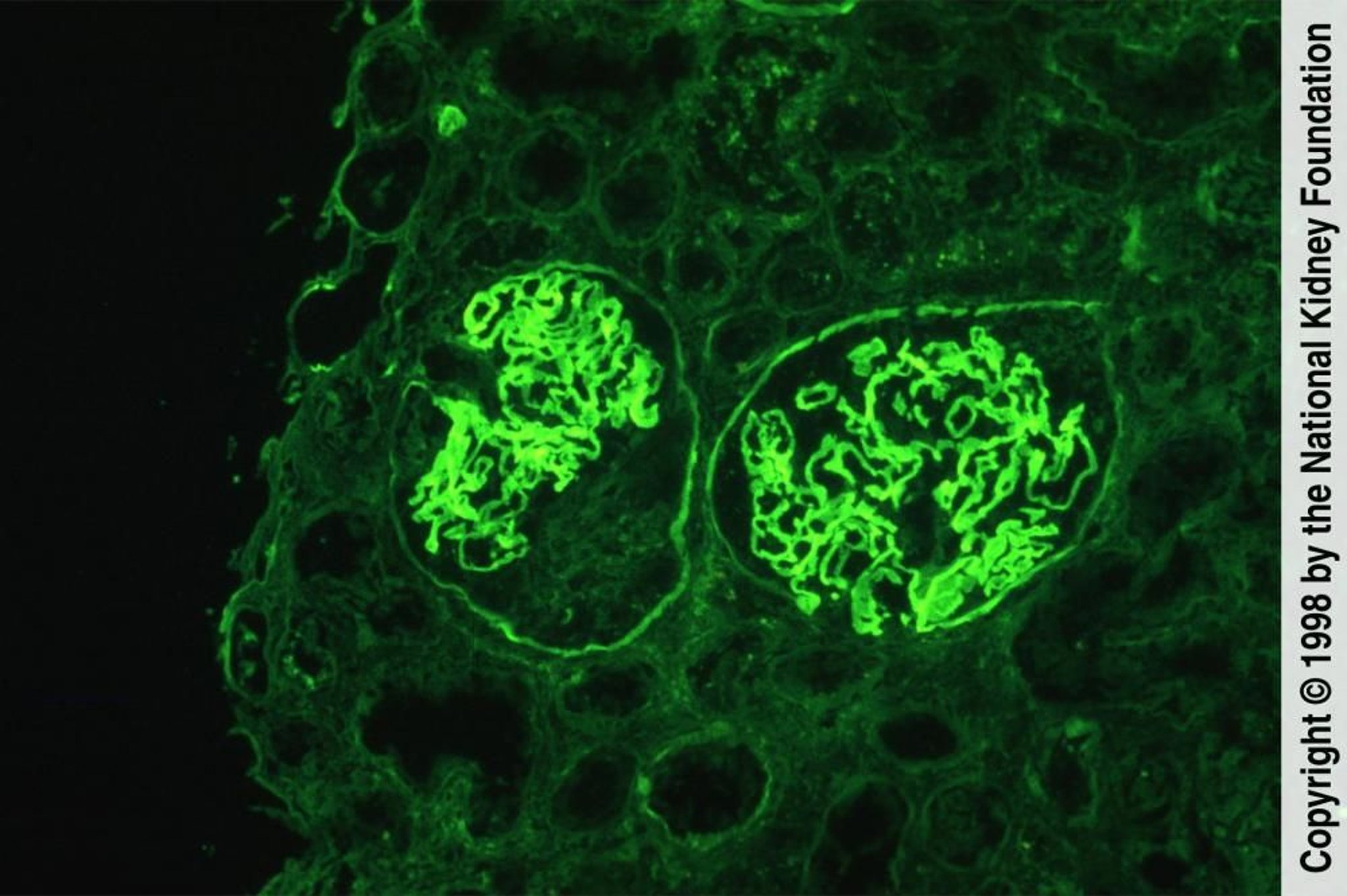
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (voir www.ajkd.org).
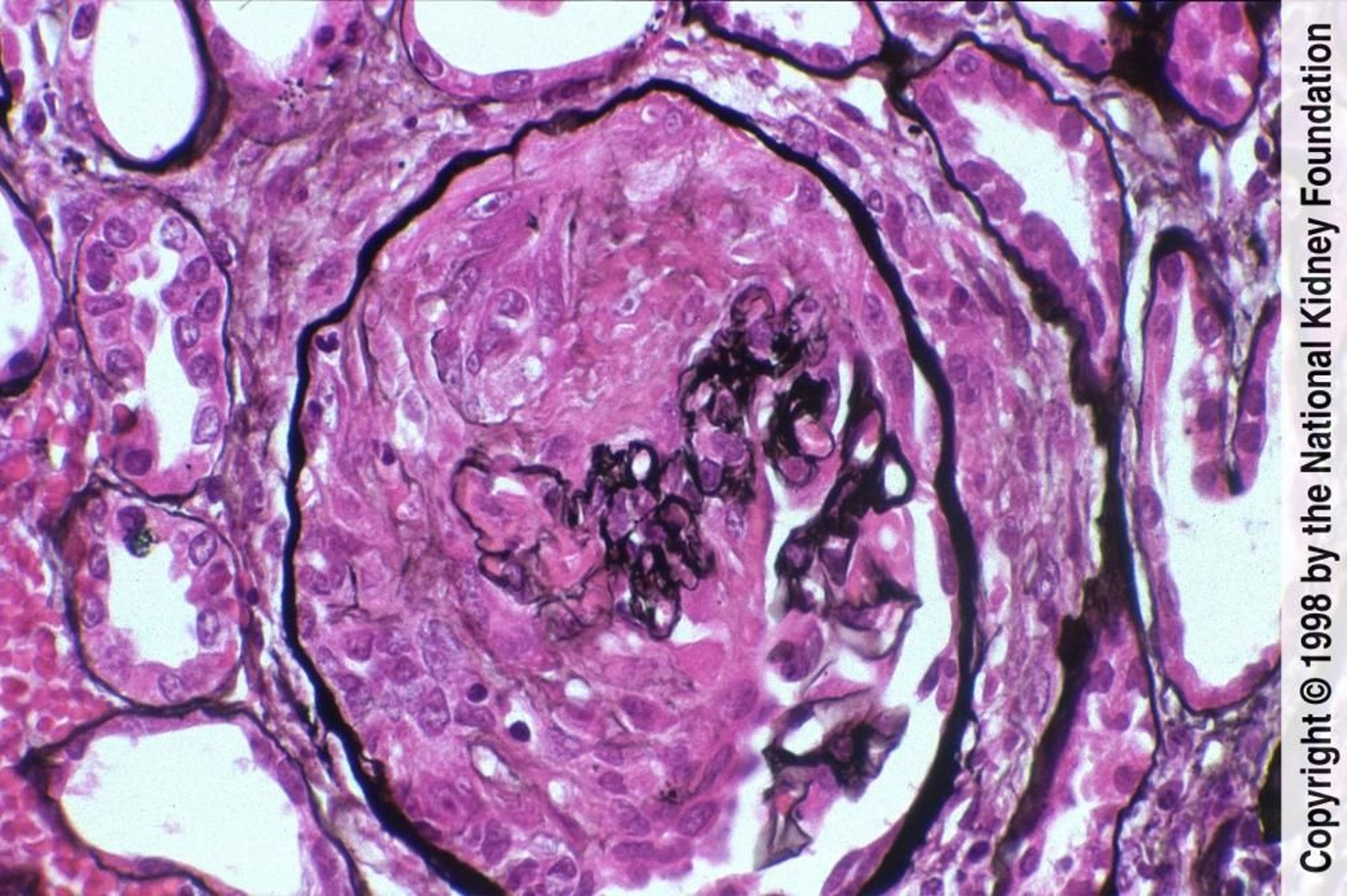
Image fournie par Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (voir www.ajkd.org).
Microscopie en immunofluorescence les signes diffèrent pour chaque type:
Dans la maladie des anticorps anti-membrane basale glomérulaire, le dépôt linéaire ou en forme de ruban d'IgG le long de la membrane basale glomérulaire est plus important et est souvent accompagné d'un dépôt linéaire et parfois granuleux de C3.
Dans les glomérulonéphrites rapidement progressives à complexes immuns, l'immunofluorescence révèle l'existence de dépôts mésangiaux diffus d'IgG et des dépôts de C3.
Dans la glomérulonéphrite rapidement progressive pauci-immunitaire, l'immunofluorescence est négative, ne détectant aucun dépôt. Cependant, les croissants contiennent de la fibrine, quel que soit l'aspect observé en immunofluorescence.
Dans la glomérulonéphrite rapidement progressive à double anticorps, la coloration linéaire de la membrane basale glomérulaire est présente.
Dans la glomérulonéphrite rapidement progressive idiopathique, certains patients présentent des complexes immuns et d'autres n'ont pas de coloration immune et de dépôts.
Traitement de la glomérulonéphrite rapidement progressive
Corticostéroïdes
Cyclophosphamide
Rituximab
Échanges plasmatiques
Le traitement varie en fonction du type de la maladie, bien qu'aucun protocole thérapeutique n'ait été rigoureusement étudié. Le traitement doit être instauré précocement, idéalement lorsque la créatininémie est < 5 mg/dL (442 micromoles/L) et avant que la biopsie ne montre une atteinte en croissant de tous les glomérules ainsi qu'une fibrose interstitielle et des tubules atrophiques. Même les patients présentant une atteinte rénale et des taux de créatinine plus élevés doivent être traités de manière agressive s'ils ne nécessitent pas un traitement de suppléance rénale immédiate. Le traitement se révèle moins efficace au fur et à mesure que ces signes deviennent plus importants, et peut être nocif chez certains patients (p. ex., personnes âgées et patients atteints d'une infection).
Des corticostéroïdes et du cyclophosphamide sont habituellement administrés. Dans le cas de la glomérulonéphrite rapidement progressive à complexes immuns et pauci-immunitaire, les corticostéroïdes (méthylprednisolone 1 g IV 1 fois/jour en 30 min pendant 3 à 5 jours, suivis de prednisone 1 mg/kg par voie orale 1 fois/jour) peuvent réduire la créatininémie ou retarder la dialyse de > 3 ans chez 50% des patients (1, 2).
Le cyclophosphamide est souvent administré et peut être particulièrement bénéfique chez les patients qui ont des ANCA (antineutrophil cytoplasmic antibody); les protocoles de bolus mensuels IV ont moins d'effets indésirables, (p. ex., leucopénie, infection) que le traitement par voie orale en raison d'un dosage cumulatif réduit. On débute habituellement la prednisone et le cyclophosphamide en même temps que les échanges plasmatiques dans la maladie à anticorps anti-MBG et on poursuit le traitement afin de minimiser la néoformation d'anticorps. Les patients qui présentent une maladie idiopathique sont habituellement traités par des corticostéroïdes et du cyclophosphamide, mais les données sur l'efficacité de ce traitement sont rares.
Le rituximab peut être dosé à 375 mg/m2 par semaine pendant 4 semaines tel qu'utilisé dans l'essai RAVE (titre officiel: Rituximab in ANCA-Associated Vasculitis; [2]). Un autre protocole est une dose initiale de 1 g suivie d'une autre dose de 1 g 2 semaines plus tard. Le rituximab n'a pas été utilisé dans le traitement de la maladie anti-MBG.
Les échanges plasmatiques (tous les jours, échanges de 3 à 4 L pendant 14 jours) sont recommandés dans la maladie à anti-membrane basale glomérulaire. Des échanges plasmatiques peuvent également être envisagés en cas de glomérulonéphrite rapidement progressive liée à des complexes immuns et de glomérulonéphrite rapidement progressive pauci-immune associée aux ANCA avec hémorragie pulmonaire ou dysfonction rénale sévère au début des troubles (créatinine sérique > 5-7 mg/dL [442 à 618,8 micromoles/L] ou dépendance à la dialyse), mais son utilisation reste controversée. Les échanges plasmatiques sont supposés soustraire rapidement les anticorps libres, les complexes immuns intacts et les médiateurs de l'inflammation (p. ex., fibrinogène, complément). Bien que certaines données suggèrent que les échanges plasmatiques améliorent les résultats rénaux à court terme, un essai randomisé ultérieur n'a pas montré qu'ils réduisaient la mortalité ou l'incidence des maladies rénales au stade terminal (3).
Un traitement immunosuppresseur agressif peut également être bénéfique chez les patients qui présentent des taux de créatinine plus élevés. La plasmaphérèse associée à la prednisone et au cyclophosphamide s'est révélée bénéfique en cas d'atteinte rénale qui ne nécessitait pas un traitement de suppléance rénale immédiat, même si les taux de créatinine étaient supérieurs à 5-7 mg/dL (442 à 618,8 micromoles/L; [4]).
La transplantation rénale est efficace dans tous les types de maladie, mais la maladie peut récidiver sur le greffon; le risque diminue avec le temps. Dans la maladie à anticorps anti-membrane basale glomérulaire, les titres d'anticorps anti-membrane basale glomérulaire doivent être indétectables depuis au moins 12 mois avant la transplantation. En cas de glomérulonéphrite rapidement progressive pauci-immune, la maladie doit être quiescente pendant au moins 6 mois avant la transplantation; les titres d'ANCA ne doivent pas nécessairement être supprimés.
Références pour le traitement
1. Ponticelli C, Altieri P, et al: A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 9(3):444, 1998. doi: 10.1681/ASN.V93444
2. Jones RB, Cohen Tervaert JW, Hauser T: Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363:211-220, 2010. doi: 10.1056/NEJMoa0909169
3. Walsh M, Merkel PA, Peh C-A, et al: Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 382(7):621-631, 2020. doi: 10.1056/NEJMoa1803537
4. Levy JB, Turner AN, Rees AJ, et al: Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 134(11):1033-1042, 2001. doi: 10.7326/0003-4819-134-11-200106050-00009
Pronostic de la glomérulonéphrite rapidement progressive
La rémission spontanée est rare et 80 à 90% des patients non traités présentent une néphropathie terminale en 6 mois. Le pronostic est meilleur en cas de traitement précoce.
Les facteurs pronostiques favorables sont une glomérulonéphrite rapidement progressive due à:
Maladie anti-membrane basale glomérulaire, si le traitement est précoce, en particulier lorsqu'elle est traitée avant que l'oligurie n'apparaisse et lorsque la créatininémie est < 7 mg/dL (618,8 micromoles/L)
Les facteurs pronostiques défavorables comprennent:
Âge > 60 ans
Insuffisance rénale oligurique
Créatininémie élevée
Croissants circonférentiels dans > 75% des glomérules
Glomérulonéphrite rapidement progressive pauci-immune
Environ 30% des patients atteints d'une glomérulonéphrite rapidement progressive pauci-immunitaire ne répondent pas au traitement; parmi ceux-ci, environ 40% ont besoin d'une dialyse et 33% décèdent dans les 4 ans. En comparaison, une dialyse est nécessaire chez < 20% des patients répondant au traitement et près de 3% d'entre eux décèdent.
Les patients qui présentent une maladie à double anticorps semblent n'avoir pas un pronostic rénal réellement meilleur que les patients qui n'ont que la maladie des anticorps anti-membrane basale glomérulaire et leur pronostic est plus mauvais que les patients qui présentent une glomérulonéphrite rapidement évolutive pauci-immunitaire.
Les patients qui guérissent d'une glomérulonéphrite rapidement progressive présentent des modifications histologiques résiduelles, principalement au niveau des glomérules, qui consistent essentiellement en une hypercellularité, avec peu ou pas de sclérose au niveau du flocculus glomérulaire ou des cellules épithéliales avec fibrose minime de l'interstitium.
Le décès est habituellement dû à des causes infectieuses ou cardiaques, en supposant qu'un décès urémique soit prévenu par la dialyse.
Points clés
Évoquer une glomérulonéphrite rapidement progressive en cas de lésions rénales aiguës avec hématurie et dysmorphie des globules rouges ou cylindres de globules rouges, en particulier en cas de symptômes non spécifiques ou constitutionnels ou subaigus (p. ex., fatigue, fièvre, anorexie, arthralgies, douleurs abdominales).
Évaluer par des tests sérologiques et une biopsie rénale précoce.
Initier un traitement précoce, par des corticostéroïdes, du cyclophosphamide et dans certains cas des échanges plasmatiques.
Envisager une transplantation rénale après avoir contrôlé l'activité de la maladie.







