







L'histiocytose pulmonaire à cellules de Langerhans se caractérise par une prolifération monoclonale de cellules de Langerhans au niveau de l'interstitium pulmonaire et des alvéoles. L'étiologie est inconnue, mais le tabagisme joue un rôle primordial. Les symptômes sont une dyspnée, une toux, une fatigue et une douleur pleurale. Le diagnostic repose sur l'anamnèse, l'imagerie médicale et parfois un lavage bronchoalvéolaire et les résultats d'une biopsie. Le traitement consiste dans le sevrage tabagique. Les corticostéroïdes sont administrés dans de nombreux cas, mais leur efficacité est douteuse. La transplantation pulmonaire est habituellement curative quand elle est associée au sevrage tabagique. La survie à 5 ans est d'environ 74%. Les patients ont un risque accru de cancer.
(Voir aussi Revue générale des maladies pulmonaires interstitielles et Histiocytoses à cellules de Langerhans.)
L'histiocytose pulmonaire à cellules de Langerhans est une maladie dans laquelle les cellules de Langerhans monoclonales CD1a positives (un type d'histiocyte) infiltrent les bronchioles et l'interstitium alvéolaire, conjointement avec des lymphocytes, des plasmocytes, des neutrophiles et des éosinophiles. L'histiocytose pulmonaire à cellules de Langerhans est une manifestation de l'histiocytose à cellules de Langerhans, qui peut affecter divers organes (notamment les poumons, la peau, les os, l'hypophyse et les ganglions lymphatiques), isolément ou simultanément. L'histiocytose pulmonaire à cellules de Langerhans affecte un seul organe dans ≥ 85% des cas.
L'étiologie de l'histiocytose pulmonaire à cellules de Langerhans est inconnue, mais la maladie est observée presque exclusivement chez les Blancs âgés de 20 à 40 ans qui fument. L'homme est autant touché que la femme. Les femmes développent la maladie plus tard, mais les différences au début liées au sexe peuvent correspondre à des différences de comportement face au tabac. La physiopathologie pourrait impliquer l'induction et la prolifération des cellules de Langerhans en réponse à des cytokines et à des facteurs de croissance sécrétés par les macrophages alvéolaires en réponse à la fumée de cigarettes.
Symptomatologie de l'histiocytose pulmonaire à cellules de Langerhans
La symptomatologie typique de l'histiocytose pulmonaire à cellules de Langerhans est une dyspnée, une toux non productive, une asthénie, une fièvre, une perte de poids et des douleurs thoraciques pleurales. Un pneumothorax soudain et spontané est fréquent.
Environ 15% des patients sont asymptomatiques, la maladie étant découverte fortuitement sur une rx thorax pratiquée pour une autre raison.
La douleur osseuse due à des kystes osseux (18%), l'éruption cutanée (13%) et la polyurie due à un déficit en arginine vasopressine (5%) sont les manifestations les plus fréquentes de l'atteinte extrapulmonaire et se produisent chez jusqu'à 15% des patients; ce sont rarement les premiers symptômes de l'histiocytose pulmonaire à cellules de Langerhans. Il existe peu de signes dans l'histiocytose pulmonaire à cellules de Langerhans; l'examen clinique est habituellement normal.
Diagnostic de l'histiocytose pulmonaire à cellules de Langerhans
TDM à haute résolution
Épreuves fonctionnelles respiratoires
Parfois, bronchoscopie et biopsie
On suspecte une histiocytose pulmonaire à cellules de Langerhans en fonction de l'anamnèse et de la rx thorax et elle est confirmée par la TDM à haute résolution et la bronchoscopie avec biopsie et lavage bronchoalvéolaire.
La rx thorax révèle habituellement des opacités nodulaires bilatérales et symétriques au niveau des segments médians et supérieurs des poumons ainsi que des modifications kystiques et des volumes pulmonaires normaux ou augmentés. Les bases pulmonaires sont souvent épargnées. L'aspect des images peut simuler une broncho-pneumopathie chronique obstructive (BPCO) ou une lymphangioléiomyomatose.
La mise en évidence à la TDM à haute résolution de kystes du lobe supérieur et des régions moyennes (souvent de formes étranges) et/ou de nodules associés à un épaississement interstitiel est considérée comme un argument diagnostique de l'histiocytose pulmonaire à cellules de Langerhans.

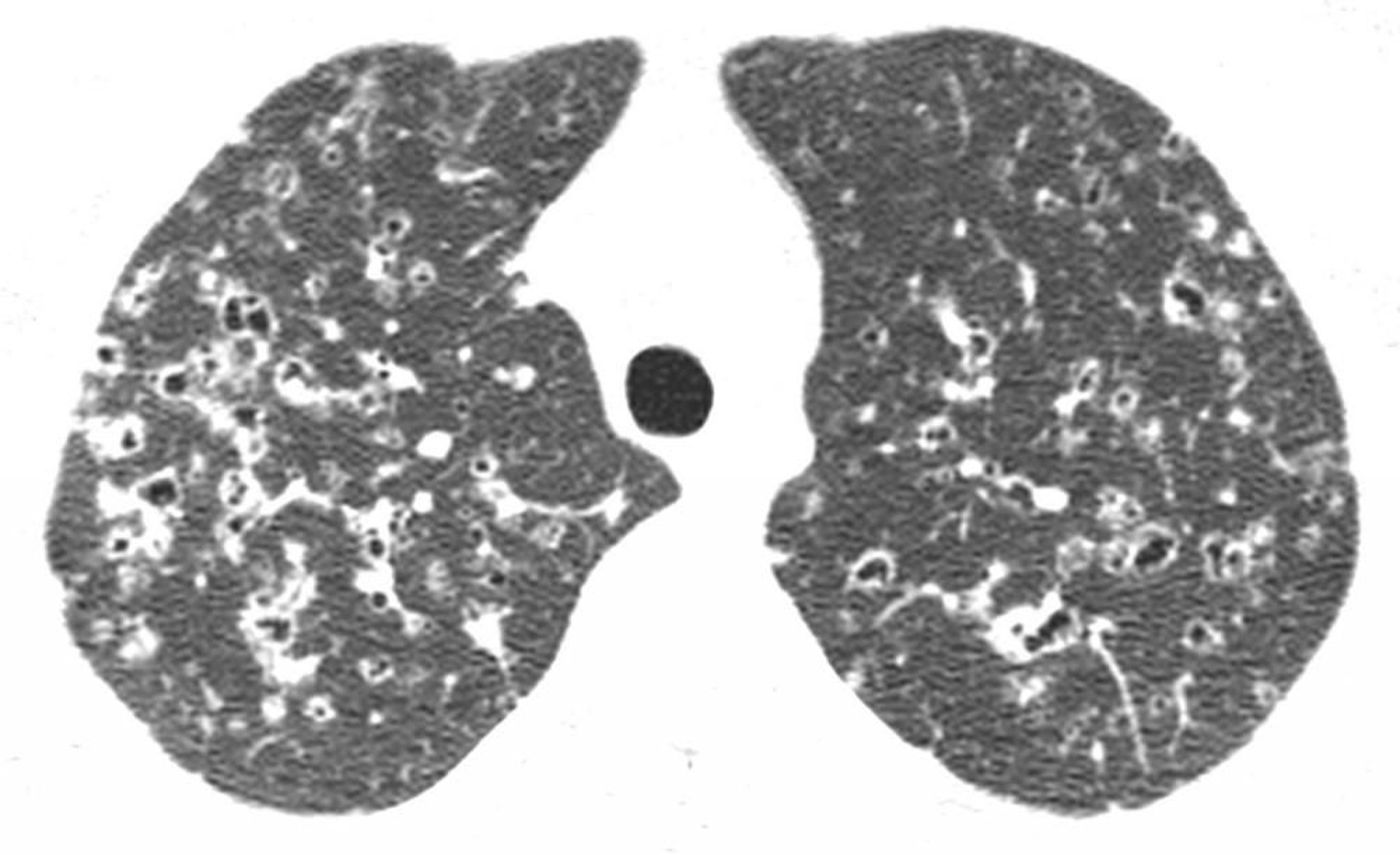
Image courtesy of Harold R. Collard, MD.
Les résultats des épreuves fonctionnelles respiratoires peuvent être normaux, montrer un trouble restrictif, obstructif ou mixte selon le stade auquel le test est effectué au cours de la maladie. Le plus souvent, la capacité de diffusion du monoxyde de carbone (DLCO) et d'effort est réduite.
Une bronchoscopie et une biopsie sont indiquées lorsque l'imagerie médicale et les épreuves fonctionnelles respiratoires ne sont pas concluantes. La découverte de > 5% de cellules CD1a dans le liquide de lavage bronchoalvéolaire est très évocatrice de la maladie. La biopsie montre une prolifération de cellules de Langerhans avec parfois un amas d'éosinophiles (l'origine du terme désuet de granulome à éosinophiles) au milieu de nodules cellulaires et fibrotiques qui peut prendre une configuration stellaire. La coloration immunohistochimique est positive pour CD1a, la protéine S-100 et les antigènes HLA-DR.
Traitement de l'histiocytose pulmonaire à cellules de Langerhans
Sevrage tabagique
Peut-être des corticostéroïdes et des médicaments cytotoxiques (vinblastine, cladribine) ou la transplantation pulmonaire
Le principal traitement de l'histiocytose pulmonaire à cellules de Langerhans consiste à arrêter le tabac, ce qui induit la disparition des symptômes dans une proportion allant jusqu'à 1/3 des patients.
L'administration empirique de corticostéroïdes et de médicaments immunosuppresseurs est une pratique fréquente bien que son efficacité ne soit pas prouvée.
La transplantation pulmonaire reste une éventualité chez les patients par ailleurs en bonne santé développant rapidement une insuffisance respiratoire, mais la maladie peut réapparaître dans le poumon transplanté si le patient continue ou se remet à fumer.
Pronostic de l'histiocytose pulmonaire à cellules de Langerhans
Une résolution spontanée des symptômes est observée chez certains patients présentant une symptomatologie d'histiocytose pulmonaire à cellules de Langerhans légère. Les taux de survie à 10 ans sont > 90% (1). Les marqueurs cliniques de la maladie évolutive comprennent
Continuer à fumer
Ages extrêmes
Atteinte multiviscérale
Symptômes constitutionnels persistants
Nombreux kystes sur la rx thorax
DLCO réduite
VEMS1 bas (VEMS1)/capacité vitale forcée (CVF) (< 66%)
Rapport volume résiduel élevé (VR)/capacité pulmonaire totale (CPT) (> 33%)
Besoin de corticostéroïdes au long cours
La cause du décès est l'insuffisance respiratoire ou un cancer induit par le tabac. Le risque de cancer du poumon est augmenté par la fumée de cigarette.
Référence pour le pronostic
1. Benattia A, Bugnet E, Walter-Petrich A, et al. Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort. Eur Respir J 2022;59(5):2101017. Published 2022 May 26. doi:10.1183/13993003.01017-2021
Points clés
Dans l'histiocytose pulmonaire à cellules de Langerhans, des cellules de Langerhans monoclonales prolifèrent dans les bronchioles et l'interstitium alvéolaire.
Évoquer une histiocytose pulmonaire à cellules de Langerhans chez les patients de 20 à 40 ans qui fument et dont la rx thorax montre des opacités nodulaires bilatérales et symétriques des champs pulmonaires moyens et supérieurs avec des anomalies kystiques.
Confirmer le diagnostic par TDM à haute résolution ou, si les résultats ne sont pas concluants, par biopsie pulmonaire.
Recommander l'arrêt du tabagisme.
Envisager des corticostéroïdes et des médicaments cytotoxiques et, si le sujet ne fume plus, la transplantation pulmonaire.







