Il linfoma di Burkitt è un linfoma non-Hodgkin a cellule B aggressivo che si manifesta nei bambini e negli adulti. Esistono forme endemiche, sporadiche e correlate all'immunodeficienza.
(Vedi anche Panoramica sul linfoma e Linfomi non-Hodgkin.)
Il linfoma di Burkitt classico è endemico nell'Africa Centrale e negli Stati Uniti costituisce il 30% dei linfomi dell'infanzia. La forma endemica in Africa spesso si presenta con un aumento di volume della mascella o delle ossa facciali.


M.A. ANSARY/SCIENCE PHOTO LIBRARY
Nel linfoma di Burkitt sporadico, predomina la malattia addominale, che spesso insorge nella regione della valvola ileocecale o del mesentere. Il tumore può causare ostruzione intestinale. Possono essere coinvolti anche siti extranodali come il cervello o altri organi solidi. Negli adulti, la malattia può raggiungere grosse dimensioni ed essere generalizzata, spesso con coinvolgimento massivo di fegato, milza e midollo osseo. Un interessamento del sistema nervoso centrale è spesso presente alla diagnosi oppure al momento della recidiva.
Il linfoma di Burkitt legato all'immunodeficienza si verifica soprattutto in pazienti con infezione da virus dell'immunodeficienza umana (HIV) e meno comunemente in pazienti sottoposti a trapianto di midollo osseo o a trapianti di organi solidi o che hanno altre cause di immunodeficienza. Nei pazienti con infezione da HIV, il linfoma di Burkitt è considerato un cancro che definisce l'AIDS.

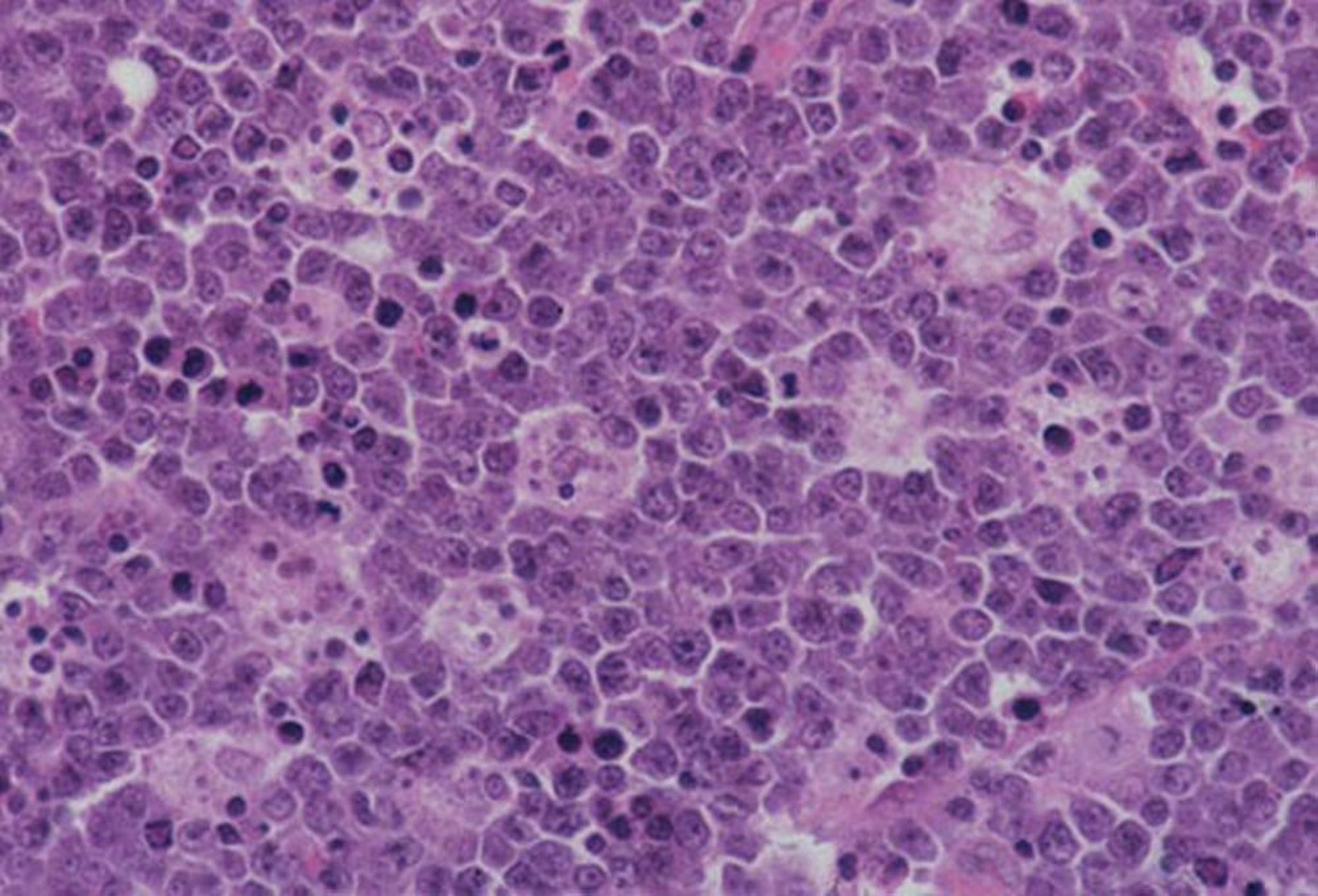
By permission of the publisher. Da Banks P, et al. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004. Disponibile all'indirizzo: www.images.md.
Il linfoma di Burkitt è la neoplasia umana a più rapido accrescimento e le valutazioni anatomopatologiche rivelano un alto tasso mitotico, una proliferazione monoclonale di cellule B e un pattern "a cielo stellato" di macrofagi benigni che hanno inglobato i linfociti maligni in apoptosi. Nelle scansioni FDG-PET (tomografia a emissione di positroni con fluorodeossiglucosio), i tumori sono altamente metabolici. Vi è una traslocazione genetica distintiva della malattia che interessa il gene c-MYC sul cromosoma 8 e la catena pesante delle immunoglobuline del cromosoma 14.
La malattia è strettamente associata all'infezione da virus di Epstein-Barr nella forma endemica; tuttavia, è incerto se il virus di Epstein-Barr abbia un ruolo eziologico.
Diagnosi del linfoma di Burkitt
Biopsia linfonodale o del midollo osseo
Raramente, laparoscopia
La diagnosi istopatologica è basata sulla biopsia di un linfonodo o del tessuto prelevato da un'altra sede sospetta di malattia, come il midollo osseo. Raramente, la laparoscopia può essere utilizzata sia per la diagnosi sia per il trattamento.
Gli esami di stadiazione devono essere accelerati perché il tumore cresce rapidamente. La stadiazione include l'imaging del tumore con fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT; se non disponibile, può essere eseguita la TC del torace, dell'addome e della pelvi.
I pazienti devono anche disporre di biopsia del midollo osseo, citologia del liquido cerebrospinale e studi di laboratorio per comprendere la lattato deidrogenasi.
Trattamento del linfoma di Burkitt
Chemioterapia intensiva
Il trattamento deve essere iniziato rapidamente in quanto questi tumori crescono rapidamente. Un regime intensivo alternato a base di ciclofosfamide, vincristina, doxorubicina, metotrexato, ifosfamide, etoposide, citarabina (CODOX-M/IVAC) associato a rituximab è utilizzato con successo nei bambini e gli adulti < 60 anni (1). Per pazienti selezionati < 60 anni di età e per molti pazienti > 60 anni, regimi come il rituximab più etoposide, prednisone, vincristina (Oncovin), e doxorubicina (R-EPOCH a dose regolata) sono comunemente utilizzati con successo. Per i pazienti senza coinvolgimento del sistema nervoso centrale, la profilassi del sistema nervoso centrale (p. es., con metotrexato sistemico e/o intratecale e/o citarabina) è essenziale.
In seguito alla terapia, è frequente la sindrome da lisi tumorale e i pazienti devono ricevere idratazione EV, allopurinolo spesso con alcalinizzazione delle urine (in assenza di iperfosfatemia) e deve essere data molta attenzione agli elettroliti (in particolare potassio, fosfato e calcio). Il rasburicase è controindicato nei pazienti con deficit di G6PD perché può causare anemia emolitica in questi pazienti. Alcuni pazienti possono richiedere la dialisi per l'iperkaliemia.
Se il paziente esordisce con ostruzione intestinale secondaria alla neoplasia ma il tumore viene completamente resecato nel corso di una laparotomia terapeutica/diagnostica, un successivo trattamento aggressivo è ancora indicato, ma possono essere necessari meno cicli. I pazienti alla fine del trattamento devono avere una risposta metabolica completa documentata dalla PET o una risposta completa documentata dalla TC e dalla biopsia del midollo osseo.
Il risultato è scarso nei pazienti nei quali l'induzione fallisce o si presenta una ricaduta (di solito nei primi 12 mesi). Devono essere considerati la terapia di salvataggio o studi clinici.
Riferimento relativo al trattamento
1. Chamuleau MED, Stenner F, Chitu DA, et al. R-CODOX-M/R-IVAC versus DA-EPOCH-R in patients with newly diagnosed Burkitt lymphoma (HOVON/SAKK): final results of a multicentre, phase 3, open-label, randomised trial. Lancet Haematol 2023;10(12):e966-e975. doi:10.1016/S2352-3026(23)00279-X
Per ulteriori informazioni
A seguire vi sono risorse in lingua inglese che possono essere utili per i medici e di aiuto e informazione per i pazienti. Si noti che il Manuale non è responsabile per il contenuto di questa risorsa.
Leukemia & Lymphoma Society: Resources for Healthcare Professionals: fornisce risorse educative per gli operatori sanitari, nonché informazioni per l'indirizzamento dei pazienti