


Torsades de pointes ventricular tachycardia is a specific form of polymorphic ventricular tachycardia in patients with a long QT interval. It is characterized by rapid, irregular QRS complexes, which appear to be twisting around the electrocardiogram (ECG) baseline. This arrhythmia may cease spontaneously or degenerate into ventricular fibrillation. It causes significant hemodynamic compromise and often death. Diagnosis is by ECG. Treatment is with IV magnesium, measures to shorten the QT interval, and direct current defibrillation when ventricular fibrillation is precipitated.
The long QT interval responsible for torsades de pointes ventricular tachycardia (TdeP VT) can be acquired, congenital or a combination.
QT-interval prolongation predisposes to early after-depolarizations and spatial dispersion of ventricular refractoriness, which can lead to arrhythmias. (For discussion of congenital causes, see Long QT Interval Syndromes, and see also Overview of Arrhythmias).
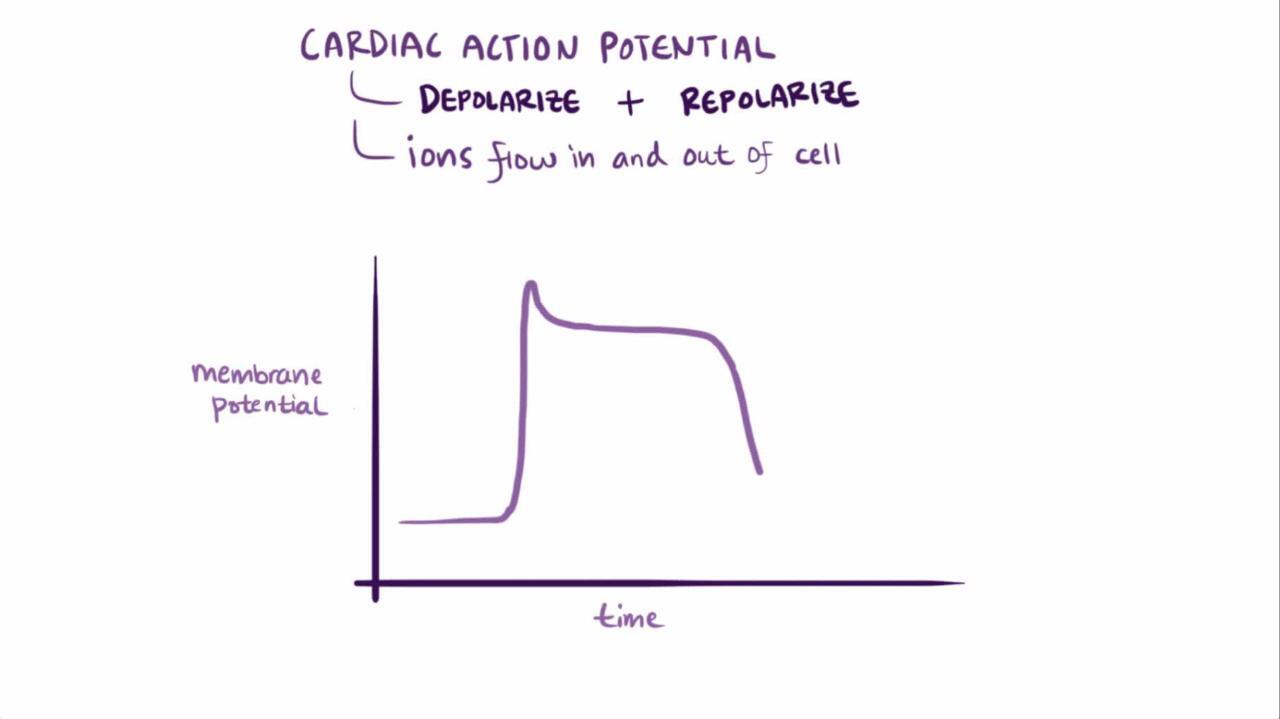
Pathophysiology of Torsades de Pointes Ventricular Tachycardia
TdeP VT results from any disorder of cardiac ion channel function or regulation that prolongs ventricular myocyte action potential duration as evidenced by prolongation of the rate-corrected QT interval on the ECG (QTc, typically calculated using Bazett's formula). The risk of TdeP VT is dependent on the degree of QTc prolongation, particularly if it is > 0.50 seconds.
TdeP VT may result from
Loss of function of repolarizing potassium current channels OR
Gain of function of depolarizing sodium or depolarizing calcium current channels
Each of these factors predisposes to TdeP VT by prolonging repolarization, which induces early after-depolarizations (secondary depolarization events during the plateau of the action potential) and spatial dispersion of ventricular refractoriness. The dispersion of refractoriness permits propagation of the early after-depolarizations and initiation of torsade de pointes ventricular tachycardia.
Predisposing factors
The most common factor in acquired TdeP VT is a
QT interval–prolonging drug, especially Class Ia, Ic, or Class III antiarrhythmic drugs
Other drugs that can induce TdeP VT include tricyclic antidepressants, phenothiazines, and certain antivirals and antifungals (see CredibleMeds for up-to-date information).
In most cases, there are several predisposing causes, which may include: female sex, older age, hypokalemia, hypomagnesemia, hypothyroidism, slow ventricular rate, irregular ventricular rate, acute intracranial disasters (eg, bleeding, stroke, traumatic brain injury), eating disorders, organophosphate poisoning, and structural heart disease (especially acute ischemia, myocarditis, and ventricular hypertrophy).
Symptoms and Signs of Torsades de Pointes Ventricular Tachycardia
TdeP VT often causes syncope because the underlying rate (200 to 250 beats/minute) is nonperfusing. Palpitations are common among conscious patients. Because the QT interval shortens with increased ventricular rates TdeP VT is often self-terminating. However, it instead may degenerate into ventricular fibrillation and cause sudden death. Sometimes the long QT interval is detected after resuscitation.
Diagnosis of Torsades de Pointes Ventricular Tachycardia
Electrocardiography (ECG)
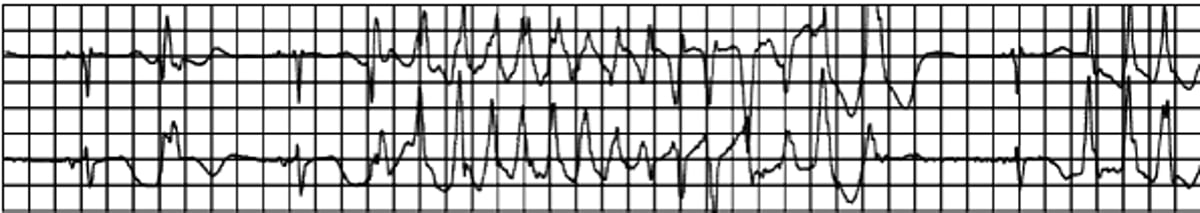
Diagnosis is by ECG showing an undulating QRS axis, with the polarity of complexes shifting around the baseline (see figure Torsades de pointes ventricular tachycardia). ECG between episodes shows a long QT interval after correction for heart rate (QTc). Normal QTc values are about 0.40 second for men and 0.41 second for women and are considered prolonged when > 0.47 second for men or > 0.48 second for women. A family history may suggest a congenital syndrome.
ECG warning signs of impending TdeP VT other than prolongation of the QT interval include
T-wave and U-wave fusion (sometimes with giant TU waves)
Post-extrasystolic changes in repolarization pattern
Macroscopic T-wave alternans (visible alternation of two different appearing T-waves)
Frequent polymorphic ventricular premature beats representing single beats of TdeP (often in bigeminy)
Repetitive short runs of polymorphic ventricular tachycardia
The TdeP VT itself is a rapid, polymorphic VT that tends to self-terminate. Onset typically follows a short-long-short RR interval initiation sequence (although this sequence is not specific for TdeP VT): The first short RR interval is between a baseline beat (usually a normal beat) and a premature beat (usually a premature ventricular beat). The long RR interval is the post-extrasystolic pause and ends with a baseline beat (usually a normal beat). The pause further prolongs the QT interval of this baseline beat and it is followed by a short RR interval when the TdeP VT begins. Although patients may have a normal QTc at other times, the QTc is usually substantially prolonged around the time of TdeP VT and the QT interval of the last QRS complex prior to TdeP VT must be long (often with a giant TU wave).
Pearls & Pitfalls
|
Torsades de pointes ventricular tachycardia

Treatment of Torsades de Pointes Ventricular Tachycardia
Unsynchronized direct current cardioversion for ventricular fibrillation
Correction of electrolyte abnormalities, especially hypokalemia
Magnesium sulfate (MgSO4) IV
Treatment of cause
An acute episode prolonged enough to cause hemodynamic compromise is treated with unsynchronized cardioversion, beginning with 100 joules. Nevertheless, early recurrence is the rule. Electrolyte abnormalities (eg, hypokalemiaantiarrhythmic drug) shortens the QT interval and may be effective especially for drug-induced torsades de pointes. Class Ia, Ic, and III antiarrhythmics are avoided.
Long-term treatment is avoidance of conditions that prolong the QT interval. Some patients with an acquired long QT-interval syndrome have an underlying subclinical congenital long QT-interval syndrome suggested by persistent QTc interval prolongation after removal of exogenous QTc prolonging influences.
Key Points
The long QT interval responsible for torsades de pointes ventricular tachycardia can be congenital or acquired.
Torsades de pointes runs are usually self-terminating but frequently recurrent.
Unsynchronized defibrillation is required if a torsades induces ventricular fibrillation.






