

Bioavailability refers to the extent and rate at which the active moiety (drug or metabolite) enters the systemic circulation, thereby accessing the site of action.
Bioavailability of a drug is largely determined by the properties of the dosage form, which depend partly on its design and manufacture. Differences in bioavailability among formulations of a given drug can have clinical significance; thus, knowing whether drug formulations are equivalent is essential.
Chemical equivalence indicates that drug products contain the same active compound in the same amount and meet current official standards; however, inactive ingredients in drug products may differ. Bioequivalence indicates that the drug products, when given to the same patient in the same dosage regimen, result in equivalent concentrations of drug in plasma and tissues. Therapeutic equivalence indicates that drug products, when given to the same patient in the same dosage regimen, have the same therapeutic and adverse effects.
Bioequivalent products are expected to be therapeutically equivalent. Therapeutic nonequivalence (eg, more adverse effects, less efficacy) is usually discovered during long-term treatment when patients who are stabilized on one formulation are given a nonequivalent substitute.
Sometimes therapeutic equivalence is possible despite differences in bioavailability. For example, the therapeutic index (ratio of the minimum toxic concentration to the median effective concentration) of penicillin is so wide that efficacy and safety are usually not affected by the moderate differences in plasma concentration due to bioavailability differences in penicillin products. In contrast, for drugs with a relatively narrow therapeutic index, bioavailability differences may cause substantial therapeutic nonequivalence.
(See also Overview of Pharmacokinetics.)
Causes of low bioavailability
Orally administered drugs must pass through the intestinal wall and then the portal circulation to the liver; both are common sites of first-pass metabolism (metabolism that occurs before a drug reaches systemic circulation). Thus, many drugs may be metabolized before adequate plasma concentrations are reached. Low bioavailability is most common with oral dosage forms of poorly water-soluble, slowly absorbed drugs.
Insufficient time for absorption in the gastrointestinal (GI) tract is a common cause of low bioavailability. If the drug does not dissolve readily or cannot penetrate the epithelial membrane (eg, if it is highly ionized and polar), time at the absorption site may be insufficient. In such cases, bioavailability tends to be highly variable as well as low.
Age, sex, physical activity, genetic phenotype, stress, disorders (eg, achlorhydria, malabsorption syndromes), or previous GI surgery (eg, bariatric surgery) can also affect drug bioavailability.
Chemical reactions that reduce absorption can decrease bioavailability. They include formation of a complex (eg, between tetracycline and polyvalent metal ions), hydrolysis by gastric acid or digestive enzymes (eg, Chemical reactions that reduce absorption can decrease bioavailability. They include formation of a complex (eg, between tetracycline and polyvalent metal ions), hydrolysis by gastric acid or digestive enzymes (eg,penicillin and chloramphenicol palmitate hydrolysis), conjugation in the intestinal wall (eg, sulfoconjugation of isoproterenol), adsorption to other drugs (eg, digoxin to cholestyramine), and metabolism by luminal microflora.
Assessing bioavailability
Bioavailability is usually assessed by determining the area under the plasma concentration–time curve (AUC—see figure Representative Plasma Concentration–Time Relationship After a Single Ora...). The most reliable measure of a drug’s bioavailability is AUC. AUC is directly proportional to the total amount of unchanged drug that reaches systemic circulation. Drug products may be considered bioequivalent in extent and rate of absorption if their plasma concentration curves are essentially superimposable.
Representative Plasma Concentration–Time Relationship After a Single Oral Dose of a Hypothetical Drug

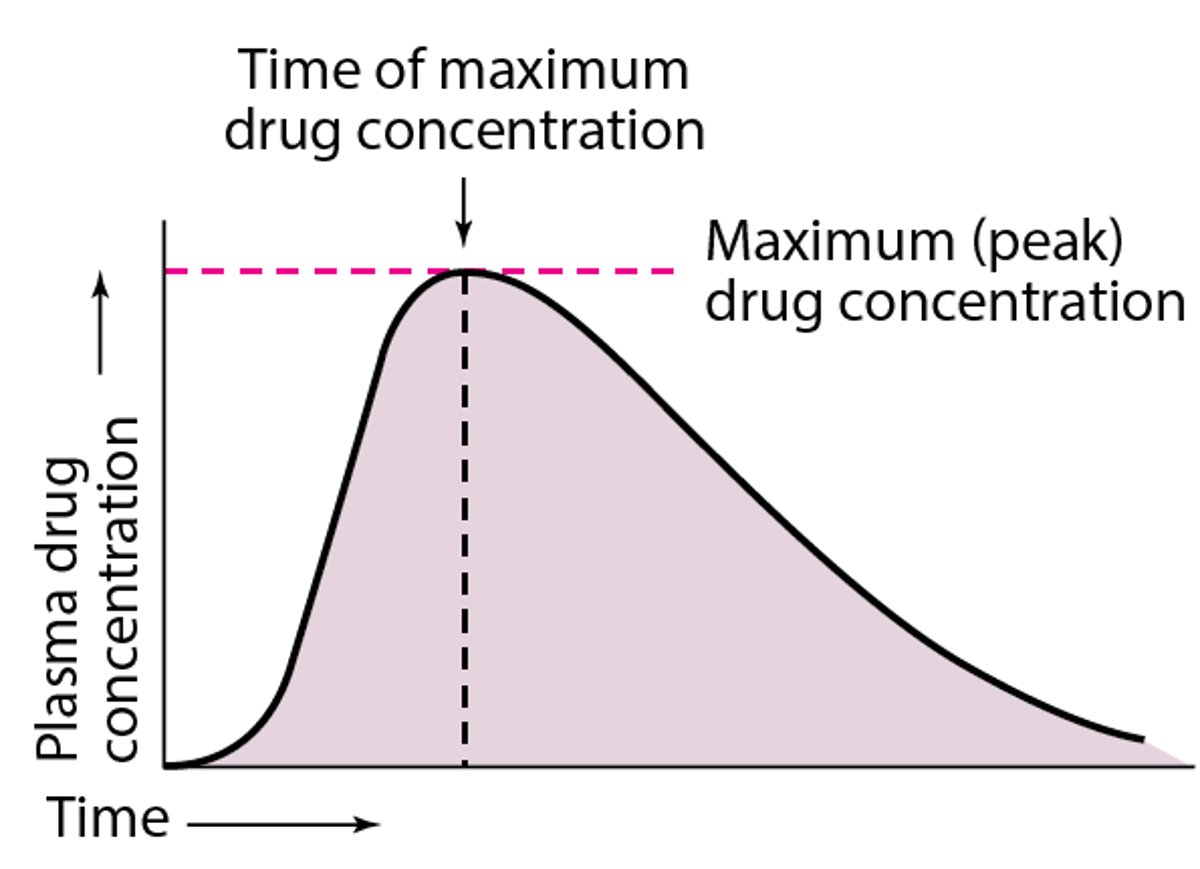
Plasma drug concentration increases with extent of absorption; the maximum (peak) plasma concentration is reached when drug elimination rate equals absorption rate. Bioavailability determinations based on the peak plasma concentration can be misleading because drug elimination begins as soon as the drug enters the bloodstream. Peak time (when maximum plasma drug concentration occurs) is the most widely used general index of absorption rate; the slower the absorption, the later the peak time.
For drugs excreted primarily unchanged in urine, bioavailability can be estimated by measuring the total amount of drug excreted after a single dose. Ideally, urine is collected over a period of 7 to 10 elimination half-lives for complete urinary recovery of the absorbed drug. After multiple dosing, bioavailability may be estimated by measuring unchanged drug recovered from urine over a 24-hour period under steady-state conditions.
Drugs Mentioned In This Article





