

Porphyria cutanea tarda (PCT) is a comparatively common hepatic porphyria affecting mainly the skin. Liver disease is also common. PCT is due to an acquired or inherited deficiency in the activity of hepatic uroporphyrinogen decarboxylase, an enzyme in the heme biosynthetic pathway (see table ). Porphyrins accumulate, particularly when there is increased oxidative stress in the hepatocytes, which is usually due to increased hepatic iron, but which may also be due to alcohol, smoking, estrogens, or hepatitis C or HIV infection. Symptoms include fragile, easily blistered skin, mainly on sun-exposed areas. Diagnosis is by porphyrin analysis of urine and stool. Differentiation from the acute cutaneous porphyrias (hereditary coproporphyria and variegate porphyria) is important. Treatment includes iron depletion by phlebotomy and enhancing porphyrin excretion by treatment with low-dose chloroquine or hydroxychloroquine. Prevention is by avoidance of sunlight, alcohol, smoking, , or hepatitis C or HIV infection. Symptoms include fragile, easily blistered skin, mainly on sun-exposed areas. Diagnosis is by porphyrin analysis of urine and stool. Differentiation from the acute cutaneous porphyrias (hereditary coproporphyria and variegate porphyria) is important. Treatment includes iron depletion by phlebotomy and enhancing porphyrin excretion by treatment with low-dose chloroquine or hydroxychloroquine. Prevention is by avoidance of sunlight, alcohol, smoking,estrogens, and iron-containing medications and with successful treatment of any concomitant hepatitis C and HIV infections.
(See also Overview of Porphyrias and Overview of Cutaneous Porphyrias.)
Pathophysiology of Porphyria Cutanea Tarda
Porphyria cutanea tarda (PCT) results from hepatic deficiency of uroporphyrinogen decarboxylase (UROD—see table ). Porphyrins accumulate in the liver and are transported to the skin, where they cause photosensitivity.
The medications that commonly trigger acute porphyria (see the Drug Database for Acute Porphyria or the American Porphyria Foundation drug database) do not trigger PCT.
Liver disease is common in PCT and may be due partly to porphyrin accumulation, chronic hepatitis C infection, concomitant hemosiderosis, or excess alcohol ingestion. Cirrhosis occurs in ≤ 35% of patients, and hepatocellular carcinoma occurs in 7 to 24% (more common among middle-aged men) (1, 2, 3).
There are 2 main types of porphyria cutanea tarda:
Type 1: Acquired or sporadic (75 to 80% of cases)
Type 2: Hereditary or familial (20 to 25% of cases)
There is also a rare type 3, which accounts for < 1% of cases.
In type 1 porphyria cutanea tarda, decarboxylase deficiency is restricted to the liver and no genetic predisposition is present. It usually manifests in middle age or later. The enzyme deficiency is thought to be acquired due to increased oxidative stress in hepatocytes that leads to increased oxidation of uroporphyrinogen to uroporphyrin, which is not a substrate for the enzyme, and to the formation of the inhibitor uroporphomethene (4). Estrogen, excess iron, and hepatitis C infection also increase such oxidative stress in hepatocytes.
In type 2 porphyria cutanea tarda, decarboxylase deficiency is inherited in an autosomal dominant fashion with limited penetrance. Deficiency occurs in all cells, including red blood cells. It may develop earlier than type 1, occasionally in childhood. The partial (~50%) deficiency in UROD activity in heterozygous patients itself is not sufficient to cause biochemical or clinical features of PCT; additional factors are required to cause the > 75% decrease in hepatic UROD activity needed for features of PCT to manifest. Such factors can include elevated hepatic iron, alcohol use, halogenated hydrocarbon exposure, hepatitis C virus or HIV infection, estrogens, and smoking. These factors increase the oxidation of uroporphyrinogens and other porphyrinogens to the corresponding porphyrins and also help form inhibitors of UROD (5).
Novel mutations in the UROD gene that contribute to development of clinical PCT, even at advanced ages (8th decade) continue to be unveiled (6).
Hepatoerythropoietic porphyria (HEP—see table ), which features profound UROD deficiency, is very rare and is often regarded as an autosomal recessive form of type 2 PCT.
Type 3 PCT, which is very rare, is hereditary but without any defect in the UROD gene; a defect in another, unidentified gene appears to be the cause.
Types 1 and 2 are the major forms of porphyria cutanea tarda. They have the same precipitants, symptoms, and treatment. Overall prevalence may be on the order of 1/10,000 but is probably higher in people exposed to halogenated aromatic hydrocarbons or other precipitants of the disease.
Pseudoporphyria
End-stage renal disease, ultraviolet radiation (UVA), and certain medications can cause PCT-like symptoms without elevated porphyrin levels (pseudoporphyria). Commonly implicated medications are furosemide, tetracyclines, sulfonamides, and naproxen and other nonsteroidal anti-inflammatory drugs (NSAIDs). End-stage renal disease, ultraviolet radiation (UVA), and certain medications can cause PCT-like symptoms without elevated porphyrin levels (pseudoporphyria). Commonly implicated medications are furosemide, tetracyclines, sulfonamides, and naproxen and other nonsteroidal anti-inflammatory drugs (NSAIDs).
Because porphyrins are poorly dialyzed, some patients receiving long-term hemodialysis develop a skin condition that resembles PCT; this condition is termed pseudoporphyria of end-stage renal disease. However, some of these patients have underlying PCT, related to some or all of the known inciting factors, including iatrogenic iron overload. It can be severe and difficult to manage because of co-existent anemia of chronic disease makes it difficult to remove iron by phlebotomy. In addition, the efficacy of hydroxychloroquine is decreased because complexes of uroporphyrin and hydroxychloroquine are formed and are not removed by hemodialysis or peritoneal dialysis. Because porphyrins are poorly dialyzed, some patients receiving long-term hemodialysis develop a skin condition that resembles PCT; this condition is termed pseudoporphyria of end-stage renal disease. However, some of these patients have underlying PCT, related to some or all of the known inciting factors, including iatrogenic iron overload. It can be severe and difficult to manage because of co-existent anemia of chronic disease makes it difficult to remove iron by phlebotomy. In addition, the efficacy of hydroxychloroquine is decreased because complexes of uroporphyrin and hydroxychloroquine are formed and are not removed by hemodialysis or peritoneal dialysis.
Pathophysiology references
1. Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med 2017;377(9):862-872. doi:10.1056/NEJMra1608634
2. Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998;27(6):1661-1669. doi:10.1002/hep.510270627
3. Egger NG, Goeger DE, Payne DA, Miskovsky EP, Weinman SA, Anderson KE. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig Dis Sci 47(2):419-426, 2002. doi:10.1023/a:1013746828074
4. Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci U S A 2007;104(12):5079-5084. doi:10.1073/pnas.0700547104
5. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012;32(6):880-893. doi:10.1111/j.1478-3231.2012.02794.x
6. Soufleris S, Moore M, Phillips JD, et al. Novel UROD mutation for porphyria cutanea tarda, type 2: a case report. AME Case Rep 2024;8:67. doi:10.21037/acr-23-66
Symptoms and Signs of Porphyria Cutanea Tarda
Patients with porphyria cutanea tarda present with chronically photo-damaged, fragile skin, mainly on sun-exposed areas. Phototoxicity is delayed, thus patients do not always connect sun exposure with symptoms.

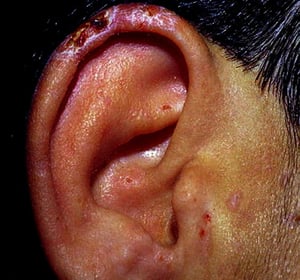
Porphyria cutanea tarda affects predominantly sun-exposed areas, including the earlobes in this patient.
Porphyria cutanea tarda affects predominantly sun-exposed areas, including the earlobes in this patient.
© Springer Science+Business Media

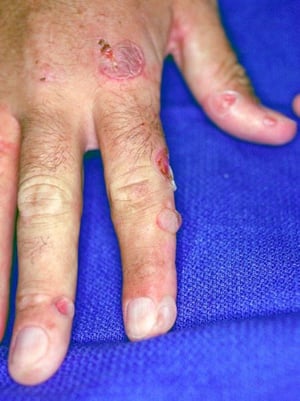
This image shows erythema and ruptured bullae on the fingers of a patient with porphyria cutanea tarda.
This image shows erythema and ruptured bullae on the fingers of a patient with porphyria cutanea tarda.
Image courtesy of Karen McKoy, MD.

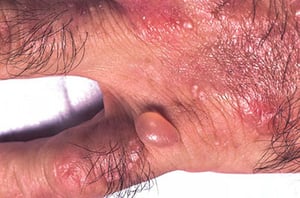
Erythema, bullae, and milia over the dorsum of the hand of a patient with porphyria cutanea tarda.
Erythema, bullae, and milia over the dorsum of the hand of a patient with porphyria cutanea tarda.
By permission of the publisher. From White K, Soter N. In Current Dermatologic Diagnosis and Treatment. Edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.

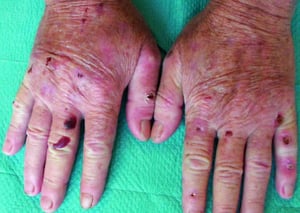
Atrophic scars have developed after rupture of bullae. Some bullae are hemorrhagic.
Atrophic scars have developed after rupture of bullae. Some bullae are hemorrhagic.
© Springer Science+Business Media
Porphyria cutanea tarda affects predominantly sun-exposed areas, including the earlobes in this patient.
Porphyria cutanea tarda affects predominantly sun-exposed areas, including the earlobes in this patient.
© Springer Science+Business Media
This image shows erythema and ruptured bullae on the fingers of a patient with porphyria cutanea tarda.
This image shows erythema and ruptured bullae on the fingers of a patient with porphyria cutanea tarda.
Image courtesy of Karen McKoy, MD.
Erythema, bullae, and milia over the dorsum of the hand of a patient with porphyria cutanea tarda.
Erythema, bullae, and milia over the dorsum of the hand of a patient with porphyria cutanea tarda.
By permission of the publisher. From White K, Soter N. In Current Dermatologic Diagnosis and Treatment. Edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.
Atrophic scars have developed after rupture of bullae. Some bullae are hemorrhagic.
Atrophic scars have developed after rupture of bullae. Some bullae are hemorrhagic.
© Springer Science+Business Media
Spontaneously or after minor trauma, tense bullae develop. Some bullae are hemorrhagic. Accompanying erosions and ulcers may develop secondary infection; they heal slowly, leaving atrophic scars, often with hypopigmentation. Sun exposure occasionally leads to erythema, edema, or itching.
Hyperemic conjunctivitis may develop, but other mucosal sites are not affected.
Areas of hypopigmentation or hyperpigmentation may develop, as may facial hypertrichosis and pseudosclerodermoid changes.
Diagnosis of Porphyria Cutanea Tarda
Elevated levels of plasma porphyrins, urinary uroporphyrin and heptacarboxyl porphyrin, and fecal isocoproporphyrin
In otherwise healthy patients, fragile skin and blister formation suggest porphyria cutanea tarda. Differentiation from acute porphyrias with cutaneous symptoms (variegate porphyria [VP] and hereditary coproporphyria [HCP]) is important because in patients with VP and HCP, the erroneous prescription of porphyrogenic medications may trigger the severe neurovisceral symptoms of the acute porphyrias. Previous unexplained neurologic symptoms or abdominal pain may suggest an acute porphyria. A history of exposure to chemicals that can cause pseudoporphyria should be sought.
Although all porphyrias that cause skin lesions are accompanied by elevated plasma porphyrins, elevated urinary uroporphyrin and heptacarboxyl porphyrin and fecal isocoproporphyrin indicate PCT. Urine levels of porphyrin precursor porphobilinogen (PBG) is normal in PCT. Urinary delta-aminolevulinic acid may be slightly increased (< 3 times the upper limit of normal). Red blood cell activity of UROD is normal in type 1 and type 3 PCT but decreased (by ~50%) in type 2.
All patients with PCT should be tested for hepatitis C and HIV infections (1). They should also be tested for iron overload with serum iron and ferritin levels, and total iron-binding capacity; if results suggest iron overload, HFE gene mutation testing for hereditary hemochromatosis should be done (2).
Diagnosis references
1. Caballes FR, Sendi H, Bonkovsky HL: Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 32(6):880–893, 2012. doi:10.1111/j.1478-3231.2012.02794.x
2. Bonkovsky HL, Guo JT, Hou W, et al: Porphyrin and heme metabolism and the porphyrias. Compr Physiol 3(1):365–401, 2013. doi:10.1002/cphy.c120006
Treatment of Porphyria Cutanea Tarda
Four complementary therapeutic strategies are available:
Reduction of body iron stores
Increase in porphyrin excretion
Treatment of chronic hepatitis C and HIV infection, when present
Cessation of alcohol use, smoking, and estrogen use, when present
These strategies can be combined for more rapid remission, although combining them is not usually necessary. The treatment is monitored by determinations of serum ferritin (if iron reduction therapy is used) and urinary porphyrin excretion every other or every third month until full remission.
Patients should stop alcohol use and smoking (including marijuana).
Iron removal by therapeutic phlebotomy is usually effective. A unit of blood is removed every week or two. When serum ferritin falls slightly below normal, phlebotomy is stopped. Usually, 6 to10 sessions are needed. Urine and plasma porphyrins fall gradually with treatment, lagging behind but paralleling the fall in ferritin. The skin eventually becomes normal. After remission, further phlebotomy is needed only if there is a recurrence. Ongoing iron reduction therapy is also recommended when hereditary hemochromatosis is present. In patients with hereditary hemochromatosis, the target serum ferritin level is 50 to 150 ng/mL (50 to 150 microgram/L).


Photos courtesy of Herbert L. Bonkovsky, MD.
Low-dose chloroquine or hydroxychloroquine, 100 to 125 mg orally twice a week, removes excess porphyrins from the liver and perhaps other tissues by increasing the excretion rate. Higher doses can cause transient liver damage and worsening of porphyria. When remission is achieved, the regimen is stopped.Low-dose chloroquine or hydroxychloroquine, 100 to 125 mg orally twice a week, removes excess porphyrins from the liver and perhaps other tissues by increasing the excretion rate. Higher doses can cause transient liver damage and worsening of porphyria. When remission is achieved, the regimen is stopped.
Chloroquine and hydroxychloroquine are not effective in advanced renal disease, and phlebotomy is usually contraindicated because of underlying anemia. However, recombinant erythropoietin mobilizes excess iron and improves the anemia enough to permit phlebotomy. In end-stage renal disease, deferoxamine is an adjunct to phlebotomy for reduction of hepatic iron, the complexed iron being removed during dialysis. Dialyzers with ultrapermeable membranes and extra high blood flow rates are needed.Chloroquine and hydroxychloroquine are not effective in advanced renal disease, and phlebotomy is usually contraindicated because of underlying anemia. However, recombinant erythropoietin mobilizes excess iron and improves the anemia enough to permit phlebotomy. In end-stage renal disease, deferoxamine is an adjunct to phlebotomy for reduction of hepatic iron, the complexed iron being removed during dialysis. Dialyzers with ultrapermeable membranes and extra high blood flow rates are needed.
Patients with overt PCT and hepatitis C infection should be evaluated for treatment with direct-acting antiviral drugs. Previous iron depletion augments the response to interferon-based antiviral therapy, but such depletion seems unnecessary when treatment is with highly effective direct-acting antiviral medications. Remission of PCT skin lesions following antiviral treatment has been reported, but documentation of complete reversal of the metabolic defects and long-term improvement is lacking (1). Nevertheless, it seems worth first treating and curing hepatitis C virus infection with direct antiviral treatment before deciding on iron reduction or hydroxychloroquine therapy in such patients (). Nevertheless, it seems worth first treating and curing hepatitis C virus infection with direct antiviral treatment before deciding on iron reduction or hydroxychloroquine therapy in such patients (2). HIV infection should be treated appropriately while treating PCT.
Children with symptomatic PCT are rarely encountered, but they are treated with small-volume phlebotomies or oral chloroquine; dosage is determined by body weight. Hepatitis C virus or HIV infection, if present, should be treated promptly.Children with symptomatic PCT are rarely encountered, but they are treated with small-volume phlebotomies or oral chloroquine; dosage is determined by body weight. Hepatitis C virus or HIV infection, if present, should be treated promptly.
Skin symptoms occurring during pregnancy are treated with phlebotomy. In refractory cases, low-dose chloroquine can be added; no teratogenic effects have been recognized. Depending on the degree of hemodilution and iron depletion, the skin symptoms usually abate as pregnancy advances.Skin symptoms occurring during pregnancy are treated with phlebotomy. In refractory cases, low-dose chloroquine can be added; no teratogenic effects have been recognized. Depending on the degree of hemodilution and iron depletion, the skin symptoms usually abate as pregnancy advances.
Pre- or postmenopausal estrogen supplementation or other estrogen therapy, such as for men with prostate cancer, is interrupted during treatment for PCT. Stopping estrogens often induces remission. Substituting transdermal estrogens for systemic estrogens is sometimes done if postmenopausal symptoms are troublesome, but some risk may remain due to some degree of systemic absorption.
Cimetidine has been suggested as a treatment of PCT, because it is believed Cimetidine has been suggested as a treatment of PCT, because it is believedcimetidine down-regulates hepatic ALA synthase-1. However, hepatic ALA synthase-1 is not measurably up-regulated in PCT. Thus, routine use of cimetidine in management of PCT is not recommended (3).
Treatment references
1. Bonkovsky HL, Rudnick SP, Ma CD, et al: Ledipasvir/Sofosbuvir Is Effective as Sole Treatment of Porphyria Cutanea Tarda with Chronic Hepatitis C. Dig Dis Sci 68(6):2738–2746, 2023. doi:10.1007/s10620-023-07859-8
2. Rudnick S, Bonkovsky HL: Hepatitis C and porphyria cutanea tarda in 2020. Aliment Pharmacol Therap 51:1432–1434, 2020. doi: 10.1111/apt.15728
3. Fujita Y, Sato-Matsumura KC: Effective treatment for porphyria cutanea tarda with oral cimetidine. J Dermatol 37(7):677–679, 2010. doi:10.1111/j.1346-8138.2010.00838.x
Prevention of Porphyria Cutanea Tarda
Patients should avoid sun exposure, using hats and clothing and zinc or titanium oxide sunscreens. Typical sunscreens that block UV light are ineffective, but UVA-absorbing sunscreens, such as those containing dibenzylmethanes, may help somewhat. Alcohol ingestion should be avoided permanently, and smoking should be stopped. If needed, estrogen supplementation, especially administered transdermally in low doses, can usually be resumed safely after a disease remission.
Key Points
Porphyria cutanea tarda (PCT) is usually acquired but may be hereditary.
Triggers include elevated hepatic iron, alcohol use, estrogen therapy, halogenated hydrocarbon exposure, hepatitis C virus infection, or HIV infection.
Medications that commonly trigger acute porphyria do not trigger PCT.
Measure urinary uroporphyrin and heptacarboxyl porphyrin, and fecal isocoproporphyrin.
Test for iron overload with serum iron and ferritin levels and total iron-binding capacity.
Test patients who have elevated serum ferritin or transferrin saturation for HFE gene mutations.
Reduce elevated iron stores by phlebotomy.
Remove excess porphyrins by giving low-dose chloroquine or hydroxychloroquine. Remove excess porphyrins by giving low-dose chloroquine or hydroxychloroquine.
Treat patients with hepatitis C virus infection to cure with direct-acting antiviral medications.
Treat patients with HIV infection with antiretroviral therapy.
More Information
The following English-language resources may be useful. Please note that The Manual is not responsible for the content of these resources.
American Porphyria Foundation: Aims to educate and support patients and families affected by porphyrias and to support research into treatment and prevention of porphyrias
American Porphyria Foundation: Safe/Unsafe Drug Database: Provides an up-to-date list of medications available in the United States to assist physicians in prescribing for patients with porphyrias
United Porphyrias Association: Provides education and support to patients and their families; provides reliable information to health-care providers; fosters and supports clinical research to improve diagnosis and management of the porphyrias
International Porphyria Network: Promotes clinical research about porphyrias
The Drug Database for Acute Porphyrias: Provides an up-to-date list of medications available in Europe to assist physicians in prescribing for patients with porphyrias
Drugs Mentioned In This Article





