

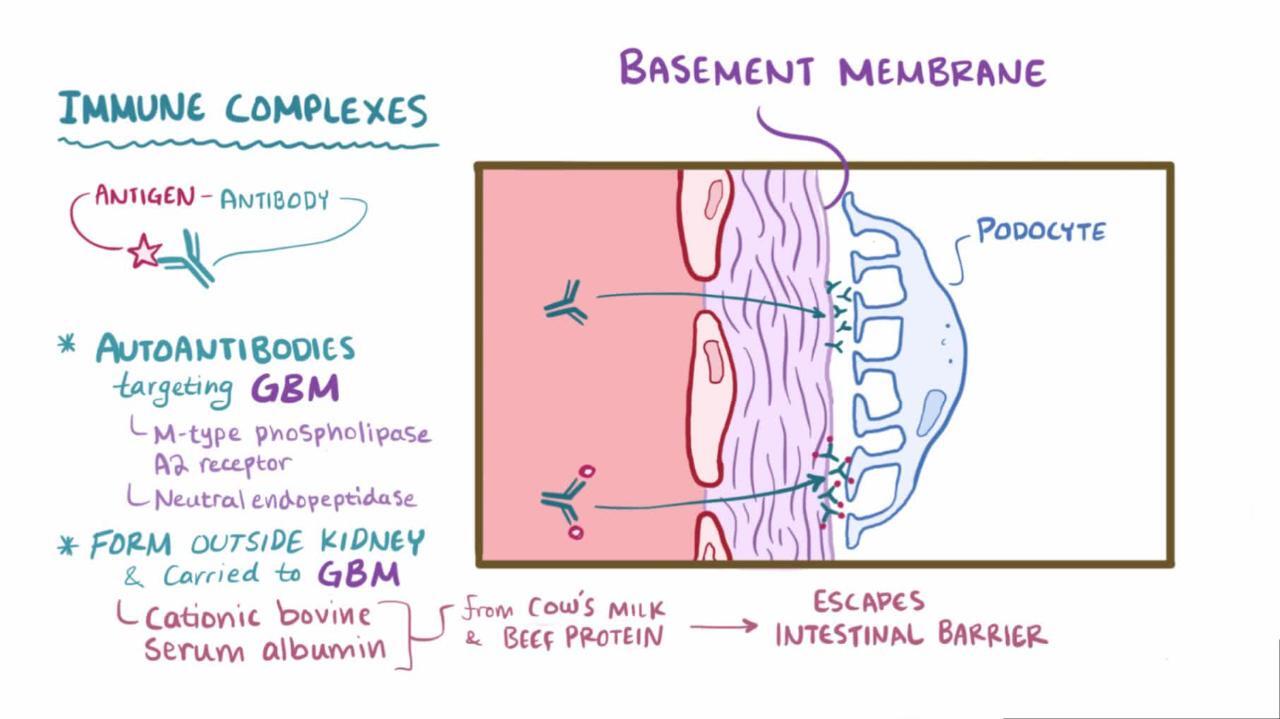
Membranous nephropathy is deposition of immune complexes on the glomerular basement membrane (GBM) with GBM thickening. Cause is usually unknown, although secondary causes include medications, infections, autoimmune disorders, and cancer. Manifestations include insidious onset of edema and heavy proteinuria with benign urinary sediment, normal kidney function, and normal or elevated blood pressure. Diagnosis is by renal biopsy. Spontaneous remission is common. Treatment of patients at high risk of progression is usually with corticosteroids and other immunosuppressive agents (eg, rituximab or cyclophosphamide).Membranous nephropathy is deposition of immune complexes on the glomerular basement membrane (GBM) with GBM thickening. Cause is usually unknown, although secondary causes include medications, infections, autoimmune disorders, and cancer. Manifestations include insidious onset of edema and heavy proteinuria with benign urinary sediment, normal kidney function, and normal or elevated blood pressure. Diagnosis is by renal biopsy. Spontaneous remission is common. Treatment of patients at high risk of progression is usually with corticosteroids and other immunosuppressive agents (eg, rituximab or cyclophosphamide).
The M-type phospholipase A2 receptor (PLA2R) in the glomerular podocyte is the major target antigen in deposited immune-complexes.
Membranous nephropathy mostly affects adults, in whom it is a common cause of nephrotic syndrome (1).

General reference
1. Alsharhan L, Beck LH Jr. Membranous Nephropathy: Core Curriculum 2021. Am J Kidney Dis 2021;77(3):440-453. doi:10.1053/j.ajkd.2020.10.009
Etiology of Membranous Nephropathy
Membranous nephropathy is usually idiopathic, but it may be secondary to any of the following:
Medications (eg, gold therapy, penicillamine, nonsteroidal anti-inflammatory drugs [NSAIDs])Medications (eg, gold therapy, penicillamine, nonsteroidal anti-inflammatory drugs [NSAIDs])
Infections (eg, hepatitis B or C virus infection, syphilis, HIV infection)
Autoimmune disorders (eg, systemic lupus erythematosus [SLE])
Thyroiditis
Cancer
Parasitic diseases (eg, malaria, schistosomiasis, leishmaniasis)
Six to 15% of patients with membranous nephropathy have an underlying cancer, including solid cancers of the lung, prostate, colon, stomach, breast, or kidney; Hodgkin or non-Hodgkin lymphoma; chronic lymphocytic leukemia; and melanoma (1).
Membranous nephropathy is rare in children and, when it occurs, is usually due to chronic hepatitis B virus infection, SLE, or autoimmune thyroid disease.
Renal vein thrombosis is more frequent in membranous nephropathy and is usually asymptomatic but may manifest with flank pain, hematuria, and hypertension. It may progress to pulmonary embolism.
Etiology reference
1. Leeaphorn N, Kue-A-Pai P, Thamcharoen N, Ungprasert P, Stokes MB, Knight EL. Prevalence of cancer in membranous nephropathy: a systematic review and meta-analysis of observational studies. Am J Nephrol 2014;40(1):29-35. doi:10.1159/000364782
Symptoms and Signs of Membranous Nephropathy
Patients typically present with edema and nephrotic-range proteinuria (≥ 3 g/day) and occasionally with microscopic hematuria and hypertension. Symptoms and signs of a disorder causing membranous nephropathy (eg, a cancer) may be present initially.
Diagnosis of Membranous Nephropathy
Renal biopsy
Evaluation for secondary causes
Diagnosis is suggested by development of nephrotic syndrome, particularly in patients who have potential causes of membranous nephropathy. The diagnosis is confirmed by renal biopsy.
Proteinuria is in the nephrotic range in 80% of patients (1). Laboratory testing is performed as indicated for nephrotic syndrome. The glomerular filtration rate (GFR), if measured, is normal or decreased.
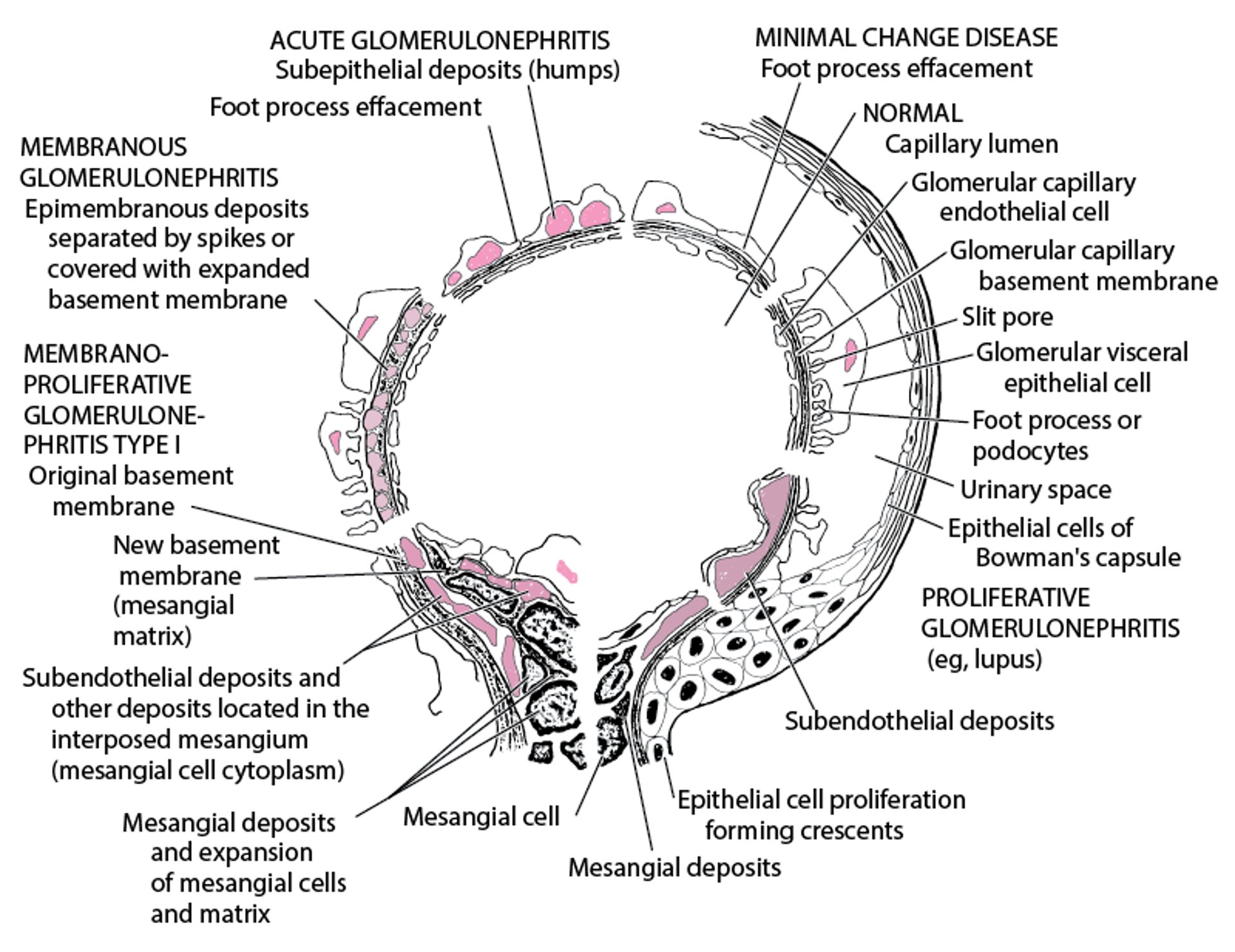
Immune complexes are seen as dense deposits on electron microscopy (see figure ). Subepithelial dense deposits occur with early disease, with spikes of lamina densa between the deposits. Later, deposits appear within the glomerular basement membrane (GBM), and marked thickening occurs. A diffuse, granular pattern of IgG deposition occurs along the GBM without cellular proliferation, exudation, or necrosis.
Identifying presence or absence of PLA2R antibody and the subclass of IgG deposits may help to differentiate idiopathic from secondary membranous nephropathy. For example, the deposits in idiopathic membranous nephropathy are PLA2R antibody positive and predominantly IgG 4, whereas PLA2R antibody is typically negative and IgG 1 and 2 predominate in malignancy-associated membranous nephropathy (2).
Electron Microscopic Features in Immunologic Glomerular Disorders


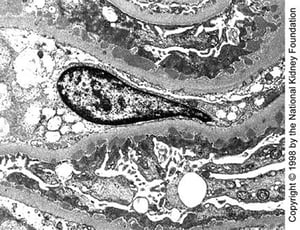
Medium-sized subepithelial dense deposits are seen on transmission electron microscopy in late stage I disease (×10,200).
Medium-sized subepithelial dense deposits are seen on transmission electron microscopy in late stage I disease (×10,200
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

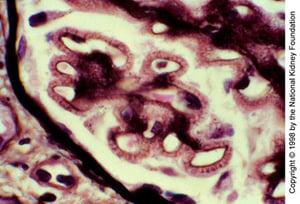
Spikes of basement membrane silver-staining material protrude from the basement membrane (high-power oil-immersion view, Jones silver stain, original magnification ×1000).
Spikes of basement membrane silver-staining material protrude from the basement membrane (high-power oil-immersion view
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

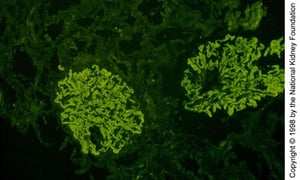
Diffuse coarsely granular IgG deposition along glomerular capillary walls (immunofluorescence with anti-IgG, original magnification ×100).
Diffuse coarsely granular IgG deposition along glomerular capillary walls (immunofluorescence with anti-IgG, original m
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Medium-sized subepithelial dense deposits are seen on transmission electron microscopy in late stage I disease (×10,200).
Medium-sized subepithelial dense deposits are seen on transmission electron microscopy in late stage I disease (×10,200
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Spikes of basement membrane silver-staining material protrude from the basement membrane (high-power oil-immersion view, Jones silver stain, original magnification ×1000).
Spikes of basement membrane silver-staining material protrude from the basement membrane (high-power oil-immersion view
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Diffuse coarsely granular IgG deposition along glomerular capillary walls (immunofluorescence with anti-IgG, original magnification ×100).
Diffuse coarsely granular IgG deposition along glomerular capillary walls (immunofluorescence with anti-IgG, original m
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Diagnosis of cause
Evaluation of patients diagnosed with membranous nephropathy usually includes the following:
A search for occult cancer, particularly in a patient who has lost weight, has unexplained anemia or heme-positive stools, or is older
Consideration of drug-induced membranous nephropathy
Antinuclear antibody testing
The search for occult cancer is usually limited to age-appropriate screening.
Diagnosis references
1. Couser WG. Primary Membranous Nephropathy [published correction appears in Clin J Am Soc Nephrol 2017 Sep 7;12(9):1528. doi: 10.2215/CJN.07190717]. Clin J Am Soc Nephrol 2017;12(6):983-997. doi:10.2215/CJN.11761116
2. Beck LH, Bonegio RG, Lambeau G: M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361(1):11, 2009. doi: 10.1056/NEJMoa0810457
Treatment of Membranous Nephropathy
Control of nephrotic-range proteinuria and/or edema with diuretics
Treatment of secondary causes of nephrotic syndrome as indicated
Immunosuppressive therapy for patients with symptomatic idiopathic membranous nephropathy at high risk of progression
Kidney transplantation for patients with kidney failure
Primary treatment is that of the causes. Among patients with idiopathic membranous nephropathy, those who are asymptomatic and have non–nephrotic-range proteinuria do not require treatment; kidney function should be monitored periodically (eg, twice yearly when apparently stable).
Patients with nephrotic-range proteinuria who are asymptomatic or who have edema should be treated with diuretics.
Patients with hypertension should be given an angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB); these medications may also benefit patients without hypertension by reducing proteinuria.
Immunosuppressive therapy
Immunosuppressants should be considered only for patients with symptomatic idiopathic membranous nephropathy and for those most at risk of progressive disease. While there is no strong evidence that immunosuppressive therapy has a long-term benefit for patient or kidney survival, it appears to improve rates of remission and possibly slows rate of progression to kidney failure (1, 2, 3). Patients who are older or chronically ill are at greater risk of infectious complications due to immunosuppressants.
No consensus protocol exists, but historically a common regimen included corticosteroids, followed by chlorambucil. However, chlorambucil is no longer preferred because of other therapeutic options with better safety profiles. Most experts favor use of a combination of rituximab and corticosteroids (No consensus protocol exists, but historically a common regimen included corticosteroids, followed by chlorambucil. However, chlorambucil is no longer preferred because of other therapeutic options with better safety profiles. Most experts favor use of a combination of rituximab and corticosteroids (2). Alternatives to rituximab include a calcineurin inhibitor and cyclophosphamide. The choice of agent is guided by disease severity and anti-PLA2R antibody levels.). Alternatives to rituximab include a calcineurin inhibitor and cyclophosphamide. The choice of agent is guided by disease severity and anti-PLA2R antibody levels.
Therapies of unproven long-term value include IV immune globulin and nonsteroidal anti-inflammatory drugs (NSAIDs).
Kidney transplantation is an option for patients with kidney failure. Membranous nephropathy recurs in approximately 30% of patients at 10 years, with loss of graft in approximately 10% (4).
Treatment references
1. von Groote TC, Williams G, Au EH, et al: Immunosuppressive treatment for primary membranous nephropathy in adults with nephrotic syndrome. Cochrane Database Syst Rev 11(11):CD004293, 2021. doi: 10.1002/14651858.CD004293.pub4
2. Fervenza FC, Appel GB, Barbour SJ, et al: Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med 381(1):36-46, 2019. doi: 10.1056/NEJMoa1814427
3. Ponticelli C, Altieri P, Scolari F, et al: A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 9(3):444, 1998. doi: 10.1681/ASN.V93444
4. Hullekes F, Uffing A, Verhoeff R, et al: Recurrence of membranous nephropathy after kidney transplantation: A multicenter retrospective cohort study. Am J Transplant 24(6):1016–1026, 2024. doi:10.1016/j.ajt.2024.01.036
Prognosis for Membranous Nephropathy
About one-third of patients undergo spontaneous remission, one-third develop stable chronic kidney disease, and one-third progress to kidney failure (1). Women, children, and young adults with non–nephrotic-range proteinuria and patients with persistently normal kidney function 3 years after diagnosis tend to have little disease progression. Patients with nephrotic-range proteinuria who are asymptomatic or who have edema that can be controlled with diuretics will have a partial or complete remission within 3 to 4 years.
Risk of progression to kidney failure is highest among patients with:
Persistent proteinuria ≥ 8 g/day, particularly men age > 50 years
An elevated serum creatinine level at presentation or diagnosis
Biopsy evidence of substantial interstitial inflammation
Prognosis reference
1. Couser WG. Primary Membranous Nephropathy [published correction appears in Clin J Am Soc Nephrol 2017 Sep 7;12(9):1528. doi: 10.2215/CJN.07190717]. Clin J Am Soc Nephrol 2017;12(6):983-997. doi:10.2215/CJN.11761116
Key Points
Membranous nephropathy is usually idiopathic; however, patients may have treatable associated disorders, such as cancers, autoimmune disorders, or infections.
Initial manifestations are typically those of nephrotic syndrome (eg, edema, nephrotic-range proteinuria, occasionally microscopic hematuria and hypertension).
Confirm the diagnosis with renal biopsy and consider associated disorders and causes.
Treat symptoms of nephrotic syndrome and treat hypertension initially with angiotensin inhibition.
Consider immunosuppressive therapy only for patients with idiopathic membranous nephropathy who are at risk for progression.
Drugs Mentioned In This Article





