



In Turner syndrome, girls are born with one of their 2 X chromosomes partly or completely missing. Diagnosis is based on clinical findings and is confirmed by cytogenetic analysis. Treatment depends on manifestations and may include surgery for cardiac anomalies and often growth hormone therapy for short stature and estrogen therapy for pubertal failure.
(See also Overview of Chromosomal Abnormalities and see Overview of Sex Chromosome Abnormalities.)
Turner syndrome is a rare genetic condition, usually occurring in phenotypic females, and is characterized by complete or partial monosomy of the X chromosome. It leads to clinical features, including short stature and ovarian dysgenesis with resultant hypergonadotropic hypogonadism and infertility, and associated multi-organ anomalies and neurocognitive deficits.
Turner syndrome occurs in approximately 1/2500 live female births worldwide (1, 2). However, 99% of 45,X conceptions abort spontaneously (3).
Approximately 45% of affected girls have a 45,X karyotype; approximately 80% have lost the paternal X. Most of the remaining 55% have mosaicism (eg, 45,X/46,XX or 45,X/47,XXX) (3). Among girls with mosaicism, the phenotype may range from that of typical Turner syndrome to normal. Occasionally, affected girls have 1 normal X and 1 X that has formed a ring chromosome. Some affected girls have 1 normal X and 1 long-arm isochromosome formed by the loss of short arms and development of a chromosome consisting of 2 long arms of the X chromosome. These girls tend to have many of the phenotypic features of Turner syndrome; thus, deletion of the X chromosome’s short arm seems to play an important role in producing the typical phenotype.
People with 1 X chromosome who have mosaicism involving a Y chromosome (ie, some cells with 46,XY) may have a male or female phenotype or even ambiguous genitalia (4).
General references
1. Martin-Giacalone BA, Lin AE, Rasmussen SA, et al: Prevalence and descriptive epidemiology of Turner syndrome in the United States, 2000-2017: A report from the National Birth Defects Prevention Network. Am J Med Genet A 191(5):1339-1349, 2023. doi: 10.1002/ajmg.a.63181
2. Nielsen J, Wohlert M: Chromosome abnormalities found among 34,910 newborn children: Results from a 13-year incidence study in Arhus, Denmark. Hum Genet 87(1):81-83, 1991. doi: 10.1007/BF01213097
3. Zhong Q, Layman LC: Genetic considerations in the patient with Turner syndrome--45,X with or without mosaicism. Fertil Steril 98(4):775-779, 2012. doi: 10.1016/j.fertnstert.2012.08.021
4. Guzewicz L, Howell S, Crerand CE, et al: Clinical phenotype and management of individuals with mosaic monosomy X with Y chromosome material stratified by genital phenotype. Am J Med Genet A 185(5):1437-1447, 2021. doi: 10.1002/ajmg.a.62127
Pathophysiology of Turner Syndrome
Common cardiac anomalies include coarctation of the aorta, aortic dilatation, and bicuspid aortic valve. Hypertension frequently occurs with aging, even without coarctation. Renal anomalies and hemangiomas are frequent. Occasionally, telangiectasia occurs in the gastrointestinal (GI) tract, with resultant GI bleeding or protein loss. Hearing loss may occur; strabismus and hyperopia (farsightedness) are common and increase the risk of amblyopia. Thyroiditis, diabetes mellitus, and celiac disease are more common than among the general population.
Infants are at higher risk of developmental dysplasia of the hip. Approximately 10% of adolescents have scoliosis. Osteoporosis and fractures are fairly common among women with Turner syndrome. Gonadal dysgenesis (ovaries replaced by bilateral streaks of fibrous stroma and devoid of developing ova) occurs in almost all patients. Between 15% and 40% of adolescents with Turner syndrome undergo spontaneous puberty, but only 2 to 10% undergo spontaneous menarche (1).
Intellectual disability is rare, but many girls have nonverbal learning disability, attention-deficit/hyperactivity disorder, or both and thus score poorly on performance tests and in mathematics, even though they score average or above in the verbal components of intelligence tests.
Pathophysiology reference
1. Zhong Q, Layman LC: Genetic considerations in the patient with Turner syndrome--45,X with or without mosaicism. Fertil Steril 98(4):775-779, 2012. doi: 10.1016/j.fertnstert.2012.08.021
Symptoms and Signs of Turner Syndrome
At and immediately after birth, neonates are usually very mildly affected; however, some present with marked dorsal lymphedema of the hands and feet and with lymphedema or loose folds of skin over the back of the neck. Other frequent anomalies include a webbed neck and a broad chest with widely spaced and inverted nipples. In childhood and adolescence, affected girls often are short in stature compared with family members, and obesity is common.
Less common findings include a low hairline on the back of the neck, ptosis, multiple pigmented nevi, short fourth metacarpals and metatarsals, prominent finger pads with whorls in the dermatoglyphics (characteristic ridges on the palms that give rise to fingerprints) on the ends of the fingers, and hypoplasia of the nails. Increased cubitus valgus (carrying angle) at the elbow occurs.


This photo shows webbed neck (frontal view) in a patient with Turner syndrome.
This photo shows webbed neck (frontal view) in a patient with Turner syndrome.
MID ESSEX HOSPITAL SERVICES NHS TRUST/SCIENCE PHOTO LIBRARY


This image shows a 13-year-old girl with typical physical characteristics of Turner syndrome such as short stature, stocky build, broad chest with widely spaced nipples ("shield" chest), lack of breast development, and cubitus valgus.
This image shows a 13-year-old girl with typical physical characteristics of Turner syndrome such as short stature, sto
By permission of the publisher. From Shulman D, Bercu B: Atlas of Clinical Endocrinology: Neuroendocrinology and Pituitary Disease. Edited by S Korenman (series editor) and ME Molitch. Philadelphia, Current Medicine, 2000.

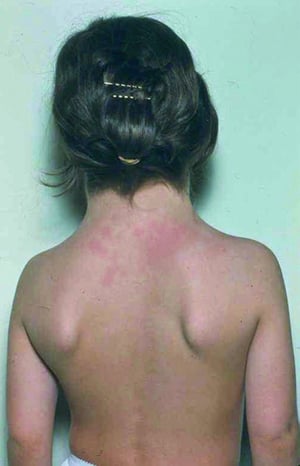
This patient with Turner syndrome has the characteristic webbed neck and low hairline.
This patient with Turner syndrome has the characteristic webbed neck and low hairline.
© Springer Science+Business Media
This photo shows webbed neck (frontal view) in a patient with Turner syndrome.
This photo shows webbed neck (frontal view) in a patient with Turner syndrome.
MID ESSEX HOSPITAL SERVICES NHS TRUST/SCIENCE PHOTO LIBRARY
This image shows a 13-year-old girl with typical physical characteristics of Turner syndrome such as short stature, stocky build, broad chest with widely spaced nipples ("shield" chest), lack of breast development, and cubitus valgus.
This image shows a 13-year-old girl with typical physical characteristics of Turner syndrome such as short stature, sto
By permission of the publisher. From Shulman D, Bercu B: Atlas of Clinical Endocrinology: Neuroendocrinology and Pituitary Disease. Edited by S Korenman (series editor) and ME Molitch. Philadelphia, Current Medicine, 2000.
This patient with Turner syndrome has the characteristic webbed neck and low hairline.
This patient with Turner syndrome has the characteristic webbed neck and low hairline.
© Springer Science+Business Media
Symptoms of cardiac anomalies depend on the extent of cardiac involvement. For instance, associated coarctation of the aorta can cause high blood pressure in the upper extremities, diminished femoral pulses, and low or absent blood pressure in the lower extremities.
Gonadal dysgenesis results in premature ovarian failure in most patients, manifested by absent or incomplete pubertal development, lack of breast development, amenorrhea (most patients have primary amenorrhea, but some have menarche and then secondary amenorrhea due to gonadal dysgenesis with premature ovarian failure), and infertility. A small proportion of patients have normal puberty and reproductive function (1). The characteristic findings are ovaries with some connective tissue with either no or few follicles (termed streak gonads) (2).
Other medical problems that are associated with Turner syndrome develop with aging and may not be evident without screening.
Symptoms and signs references
1. Pasquino AM, Passeri F, Pucarelli I, et al: Spontaneous pubertal development in Turner's syndrome. Italian Study Group for Turner's Syndrome. J Clin Endocrinol Metab 82(6):1810-1813, 1997. doi: 10.1210/jcem.82.6.3970
2. Steiner M, Saenger P: Turner Syndrome: An Update. Adv Pediatr 69(1):177-202, 2022. doi: 10.1016/j.yapd.2022.03.004
Diagnosis of Turner Syndrome
Physical examination
Cytogenetic testing by karyotyping, fluorescent in situ hybridization (FISH) analysis, and/or chromosomal microarray analysis
Testing for associated conditions
Appropriate management of Turner Syndrome varies by age and involves multidisciplinary coordinated care (1). In neonates, the diagnosis of Turner syndrome may be suspected based on the presence of lymphedema or a webbed neck. In the absence of these findings, some children are diagnosed later, based on short stature, lack of pubertal development, and amenorrhea.
Diagnosis is confirmed by cytogenetic analysis (karyotyping, FISH analysis, and/or chromosomal microarray analysis). (See also diagnosis of chromosomal abnormalities.)
Echocardiography or MRI is indicated to detect cardiac anomalies and for ongoing surveillance.
Cytogenetic analysis and Y-specific probe studies are done for all people with gonadal dysgenesis to rule out mosaicism with a Y-bearing cell line (eg, 45,X/46,XY). These people are usually phenotypic females who may have variable features of Turner syndrome. Because they are at an increased risk of gonadal tumors, especially gonadoblastomas, some of which may become malignant, prophylactic gonad removal, although controversial, is often recommended.
Concomitant medical conditions
Regular follow-up with a multidisciplinary team is recommended. Certain routine evaluations help identify problems that may be associated with Turner syndrome (2):
Cardiology evaluation with echocardiography and ECG at time of diagnosis, and ongoing evaluation to monitor for conduction abnormalities and aortic dilation
Routine monitoring for dyslipidemia and hypertension
Renal ultrasound at time of diagnosis, annual urinalysis, blood urea nitrogen, and creatinine for patients with renal system anomalies
Hearing evaluation by an audiologist and audiogram every 3 to 5 years
Evaluation for scoliosis/kyphosis annually during childhood and adolescence
Ophthalmology evaluation at age 12 to 18 months or at time of diagnosis; annual evaluation thereafter
Thyroid function tests at diagnosis and annually thereafter
Celiac screen (eg, endomysial antibody levels)
Annual screening for glucose intolerance starting at age 10 years
Close monitoring of growth and pubertal development
Diagnosis references
1. Fiot E, Alauze B, Donadille B, et al. Turner syndrome: French National Diagnosis and Care Protocol (NDCP; National Diagnosis and Care Protocol). Orphanet J Rare Dis. 2022;17(Suppl 1):261. Published 2022 Jul 12. doi:10.1186/s13023-022-02423-5
2. Shankar RK, Backeljauw PF. Current best practice in the management of Turner syndrome. Ther Adv Endocrinol Metab. 2018;9(1):33–40. doi:10.1177/2042018817746291
Treatment of Turner Syndrome
Management of comorbid conditions
Possible surgical repair of cardiac anomalies
Sometimes growth hormone and estrogen
There is no specific treatment for the underlying genetic condition. Management is based on the patient's findings.
Comprehensive management must include regular screening and management of hypertension, dyslipidemia, glucose intolerance, and thyroid dysfunction.
Coarctation of the aorta is usually repaired surgically. Other cardiac anomalies are monitored and repaired as needed.
Lymphedema can usually be controlled with support hosiery and other techniques such as massage (to improve lymph circulation and reduce undue accumulation).
Treatment with growth hormone can stimulate growth. Estrogen therapy is usually needed to initiate puberty and is typically given at age 12 to 13 years. Thereafter, estrogen/progestin contraceptives are given to maintain secondary sexual characteristics. Growth hormone can be given with estrogen therapy until epiphyses are fused (1). Continuation of estrogen therapy helps establish optimal bone density and skeletal development. Estrogen therapy may also improve the girl’s ability to plan tasks, pay attention, and assess visual and spatial relationships (2).
People with mosaic Turner syndrome may become pregnant, so they should use contraception if they are sexually active and want to prevent pregnancy. Fertility may be limited, so if they wish to become pregnant, they usually require alternative fertility interventions.
Providing neurocognitive educational and family-based psychosocial support is crucial to improve the quality of life of patients and their family members. Care of people with Turner syndrome and their families should also include genetic counseling.
Treatment references
1. Ross JL, Quigley CA, Cao D, et al. Growth hormone plus childhood low-dose estrogen in Turner's syndrome. N Engl J Med. 2011;364(13):1230-1242. doi:10.1056/NEJMoa1005669
2. Klein KO, Rosenfield RL, Santen RJ, et al. Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. J Clin Endocrinol Metab. 2018;103(5):1790-1803. doi:10.1210/jc.2017-02183
Key Points
Girls are missing all or part of 1 of their 2 X chromosomes in some or all cells tested.
Manifestations vary, but short stature, webbed neck, broad chest, gonadal dysgenesis, and cardiac anomalies (commonly coarctation of the aorta and bicuspid aortic valve) are common; intellectual disability is rare.
Risk of gonadal cancer is increased; it is often recommended to remove the gonads prophylactically, although this is controversial.
Do routine age-specific screening to detect associated medical conditions (eg, cardiac and renal anomalies).
Treat with hormones to initiate puberty and maintain secondary sexual characteristics.
Treat specific manifestations, and provide social and educational support and genetic counseling.





