





Biliary atresia is a birth defect in which the bile ducts progressively narrow and become blocked after birth, preventing bile from reaching the intestine.
This defect causes bile to collect in the liver and can lead to irreversible liver damage.
Typical symptoms include a yellowish discoloration of the skin and whites of the eyes (jaundice), dark urine, pale stools, and an enlarged liver.
The diagnosis is based on blood tests, ultrasound, radionuclide scanning, and examination of the liver and bile ducts during surgery.
Surgery is needed to create a path for bile to drain from the liver.
Bile, a digestive fluid secreted by the liver, carries away the liver's waste products and helps digest fats in the small intestine. Bile ducts carry bile from the liver to the intestine.
Biliary atresia starts several weeks to months after birth. In this defect, the bile ducts progressively narrow and become blocked. Thus, bile cannot reach the intestine. It eventually accumulates in the liver and then escapes into the blood, causing a yellowish discoloration of the skin and whites of the eyes (jaundice). Progressive, irreversible scarring of the liver, called cirrhosis, can develop as early as 2 months of age and progresses if the defect is not treated.

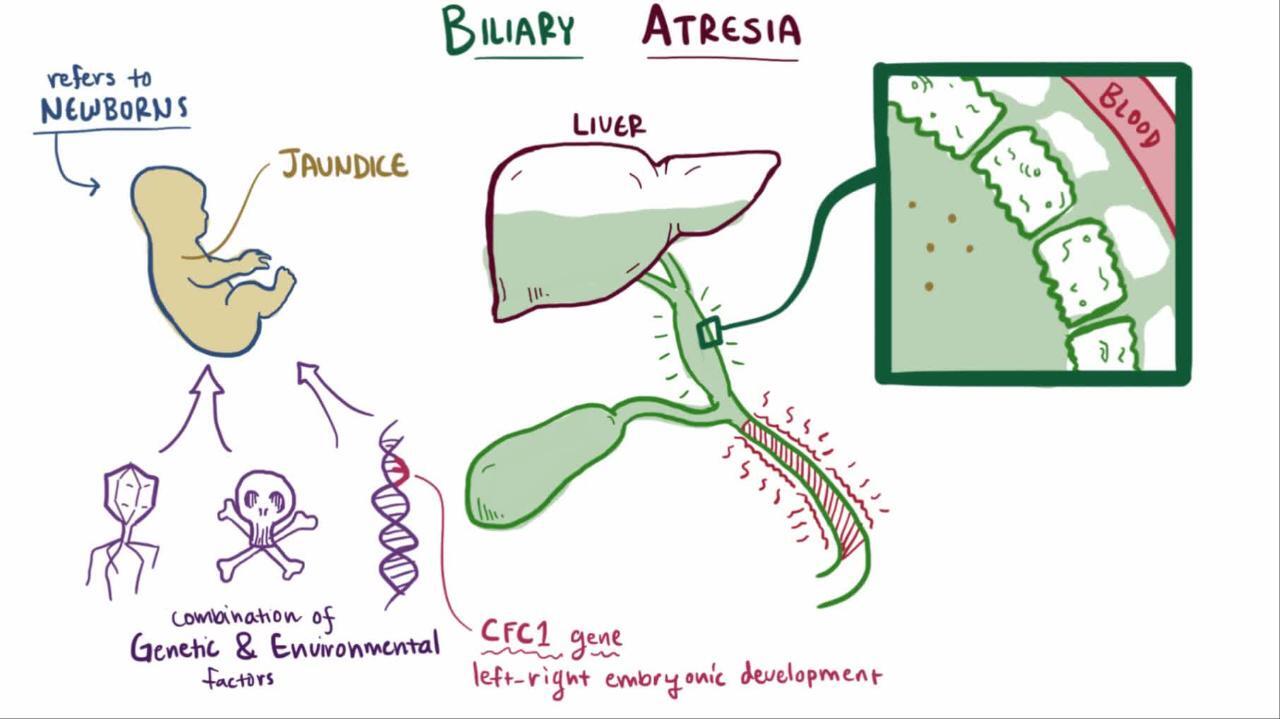
Doctors do not know why biliary atresia develops, but some infection-causing organisms and gene defects may be involved. Many infants with biliary atresia have other birth defects.
(See also Overview of Digestive Tract Birth Defects.)
Symptoms of Biliary Atresia
Infants with biliary atresia have jaundice and often have dark urine, pale stools, and a large liver and spleen. Jaundice that develops in an otherwise healthy infant 2 weeks after birth should be evaluated by a health care professional.
By the time untreated infants are 2 to 3 months old, they may not have gained weight and grown as expected. They may be itchy and irritable and have large veins visible on their abdomen, as well as a large spleen.
Diagnosis of Biliary Atresia
Blood tests
Radionuclide scanning
Ultrasounds
Liver biopsy and cholangiogram
To diagnose biliary atresia, doctors do a series of blood tests.
Doctors also do an imaging test called hepatobiliary scanning (a type of radionuclide scanning). In this test, a radioactive tracer is injected into the infant's arm, and a special scanner tracks the flow of the tracer from the liver into the gallbladder and small intestine.
Doctors may also do ultrasounds of the abdomen to further assess the liver and gallbladder.
To confirm the diagnosis, doctors do a liver biopsy and a cholangiogram. For the biopsy, doctors surgically remove a sample of tissue from the liver to test and examine it. For the cholangiogram, doctors inject a contrast agent directly into the bile ducts during surgery, which highlights the ducts on x-rays. This allows doctors to identify blockages or other abnormalities.
Treatment of Biliary Atresia
Surgery
Frequently liver transplantation
Surgery is needed to create a path for bile to drain from the liver. This operation is called a portojejunostomy, or Kasai procedure. In a portojejunostomy, the path is made by sewing a loop of intestine to the liver where the bile duct comes out. This operation should be done in the first month of life, before the liver has become scarred (cirrhosis).
After the operation, infants are often given antibiotics for a year to prevent inflammation of the bile ducts. They may also be given a medication called ursodiol. Ursodiol increases the flow of bile, which helps keep the bile drainage path open. Because good nutrition is important, infants are also given supplemental fat-soluble vitamins (vitamins A, D, E, and K).
If the infant's bile still does not flow properly 3 or more months after the operation (or if the infant cannot have the operation), the infant may need a liver transplant.
Prognosis for Biliary Atresia
Biliary atresia gets progressively worse. If untreated, it causes irreversible scarring of the liver (cirrhosis) by the time the infant is several months of age, then liver failure and death by 1 year of age.
The operation for biliary atresia does not work at all for some infants and only relieves symptoms in most infants. After the operation, 30% of people need a liver transplant when they are about 6 years old, whereas up to 80% of people need a liver transplant by the time they are 20 years old. About 80% of people survive for a long time after a liver transplant.





