



Epidermolysis bullosa is a group of 4 very rare genetic diseases and their subtypes. Epithelial fragility and easy blistering of skin and mucous membranes usually manifest at birth or in infancy. Disease phenotypes vary from mild to life-threatening. Diagnosis is by skin biopsy with immunofluorescence testing or transmission electron microscopy, and gene analysis. Treatment is supportive, and specialized multidisciplinary participation is required for severe cases.
Epidermolysis bullosa occurs in 20/million live births (1). There is no gender predilection. Occasionally, epidermolysis bullosa is not recognized until adolescence or early adulthood.
Epidermolysis bullosa is a group of inherited disorders that involve various genetic mutations. This group of disorders is distinct from epidermolysis bullosa acquisita, which is thought to be an autoimmune disease but is not inherited.
General reference
1. Fine JD: Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol 152(11):1231-1238, 2016. doi: 10.1001/jamadermatol.2016.2473
Pathophysiology of Epidermolysis Bullosa
Genetically mediated defects in epithelial adhesion proteins result in skin and mucous membrane fragility, which predisposes the epithelium to easy bullae formation after minor trauma or sometimes spontaneously. In dystrophic epidermolysis bullosa, mutations occur in the type VII collagen gene.
Types of epidermolysis bullosa
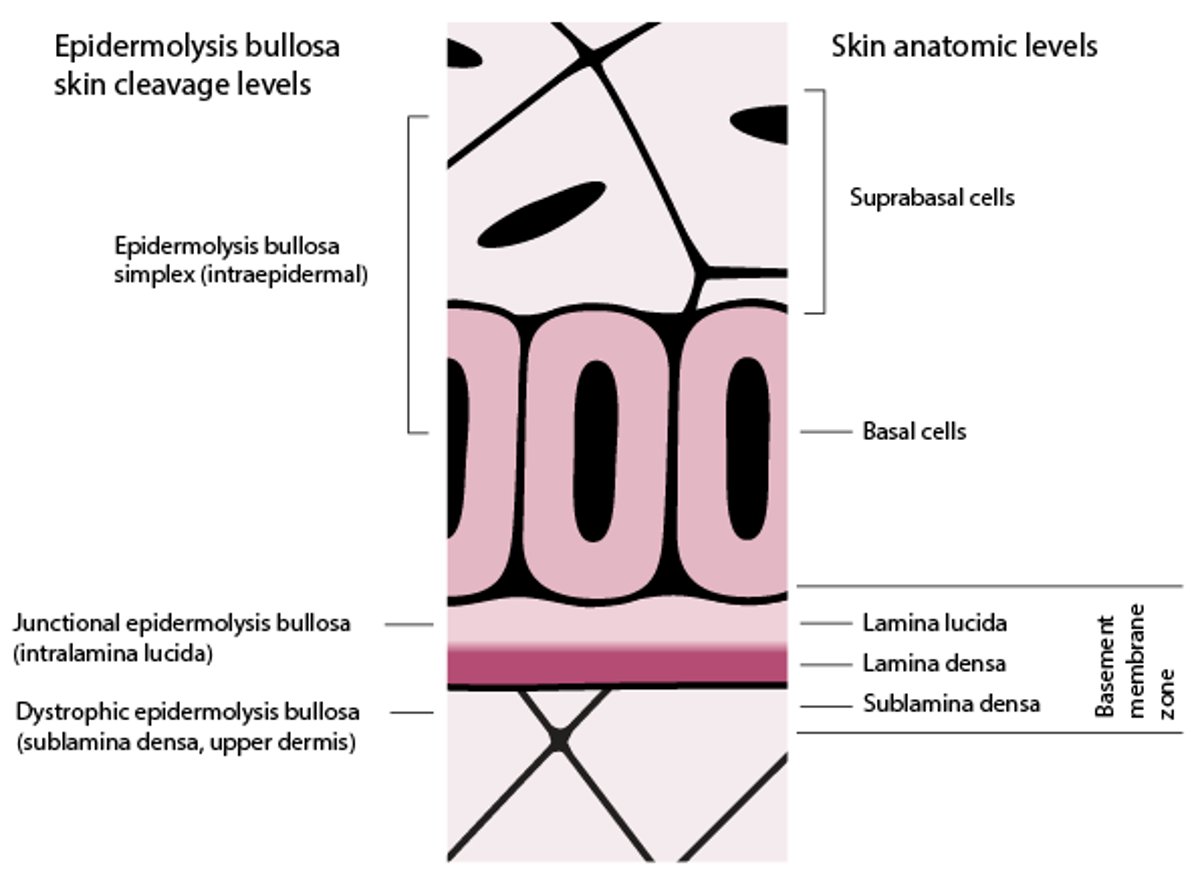
Four major epidermolysis bullosa types are defined, based on the level or levels of tissue cleavage and bullae formation (see figure Skin Cleavage Levels in Epidermolysis Bullosa) in relation to the skin’s basement membrane zone (dermal–epidermal junction):
Epidermolysis bullosa simplex: Epidermis
Junctional epidermolysis bullosa: Lamina lucida of the basement membrane zone
Dystrophic epidermolysis bullosa: Sublamina densa (uppermost dermis, just below the lamina densa of the basement membrane zone)
Kindler syndrome: Variable—intraepidermal or subepidermal
Skin Cleavage Levels in Epidermolysis Bullosa
In this figure, the basement membrane zone is disproportionately enlarged to display its layers. Epidermolysis bullosa (EB) simplex blisters form in the levels of the epidermis. Junctional EB blisters form in the lamina lucida. Dystrophic EB blisters form in the sublamina densa (uppermost dermis). Kindler syndrome blisters can form in the intraepidermal or subepidermal level. |

Different types have different inheritance patterns. (See table .)
Epidermolysis bullosa simplex is the most common and mildest type, occurring in about 80% of cases.
Multiple subtypes are defined, based on features such as
Distribution of lesions (localized vs generalized)
Relative cutaneous vs extracutaneous severity
Other skin findings (eg, exuberant granulation tissue, mottled pigmentation, pseudosyndactyly [fusion of skin between digits])
Mode of inheritance (autosomal dominant vs autosomal recessive)
Specific genes involved
Some Features of the Major Epidermolysis Bullosa Types
Type | Mode of Inheritance | Skin Cleavage Level | Characteristic Mutated Genes |
|---|---|---|---|
Epidermolysis bullosa simplex | Autosomal dominant Rarely autosomal recessive | Epidermis | KRT5, KRT14 |
Junctional epidermolysis bullosa | Autosomal recessive | Lamina lucida of the basement membrane zone | LAMB3, COL17A1, LAMC2, and LAMA3 |
Dystrophic epidermolysis bullosa | Autosomal dominant/autosomal recessive | Sublamina densa (uppermost dermis, just below the lamina densa of the basement membrane zone) | COL7A1 |
Kindler syndrome | Autosomal recessive | Variable—intraepidermal or subepidermal | FERMT1 |
Symptoms and Signs of Epidermolysis Bullosa
All types of epidermolysis bullosa manifest with painful, inappropriate blistering. Severity of symptoms correlates to severity of blistering and scarring and ranges from mild to severe.
Extensive mucocutaneous epidermolysis bullosa of any type can cause severe pain. Widespread skin lesions cause fluid imbalances and protein wasting. Skin lesions may become infected, and infections can become systemic. Mucous membrane involvement may cause malnutrition and failure to thrive, impaired breathing, and genitourinary problems.

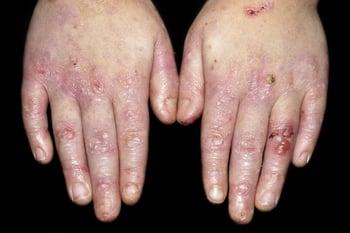
This photo shows the hands of a 5-year-old child with scars, bullae, and loss of fingernails.

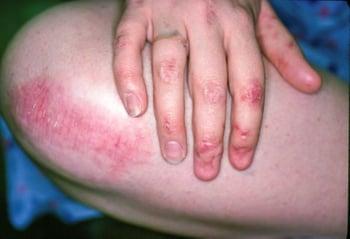
This photo shows epithelial fragility of nails and high-friction sites on the hand and knee resulting from epidermolysis bullosa.
Epidermolysis bullosa simplex
A mild subtype causes local blistering of only the palms and soles.
A severe, generalized subtype causes blistering of the trunk, arms, neck, palms, soles, and the oral mucosa may be involved.
Bullae usually heal without scarring because they lie superficially in the epidermis. Over time, hyperkeratosis develops on the palms and soles, either as focal calluses or as diffuse thickening in severe cases.
Junctional epidermolysis bullosa
A mild subtype affects only the elbows, hands, knees, and feet (typical friction sites) and often abates after infancy. Enamel hypoplasia, nail malformations, and alopecia also occur. Healing does not involve extensive scarring.
A severe, generalized mucocutaneous blistering may involve large areas of skin and also the conjunctiva, mouth, and gastrointestinal, respiratory, and genitourinary tracts.
Characteristic red, moist skin plaques (healing by granulation tissue formation) around the mouth and central face are pathognomonic. Onset is usually at birth and variants can be fatal by age 2 years.
Severe bullae heal with scarring because they occur in deeper junctional and dermal levels. Scarring of luminal lesions may cause symptomatic strictures. For example, laryngotracheal stenosis can cause stridor or a weak or hoarse cry, which implies internal involvement and a poor prognosis. Scarring of skin lesions can result in joint contractures and pseudosyndactyly.
Dystrophic epidermolysis bullosa
Mild and moderate subtypes primarily affect only the elbows, hands, knees, and feet. Nail malformations often occur and may be the sole symptom in mild cases.
A severe subtype manifests with diffuse mucocutaneous blistering at birth. The entire skin surface and the oral and gastrointestinal mucosa may be affected. Often, wide areas of skin are absent, sloughed during birth. Scarring causes a variety of external and internal complications. Pseudosyndactyly occurs nearly always.
Mild bullae heal with nonsevere scarring (worse in the moderate form) and milia (superficial epidermal inclusion cysts). Severe bullae heal with scarring because they occur in deeper levels.

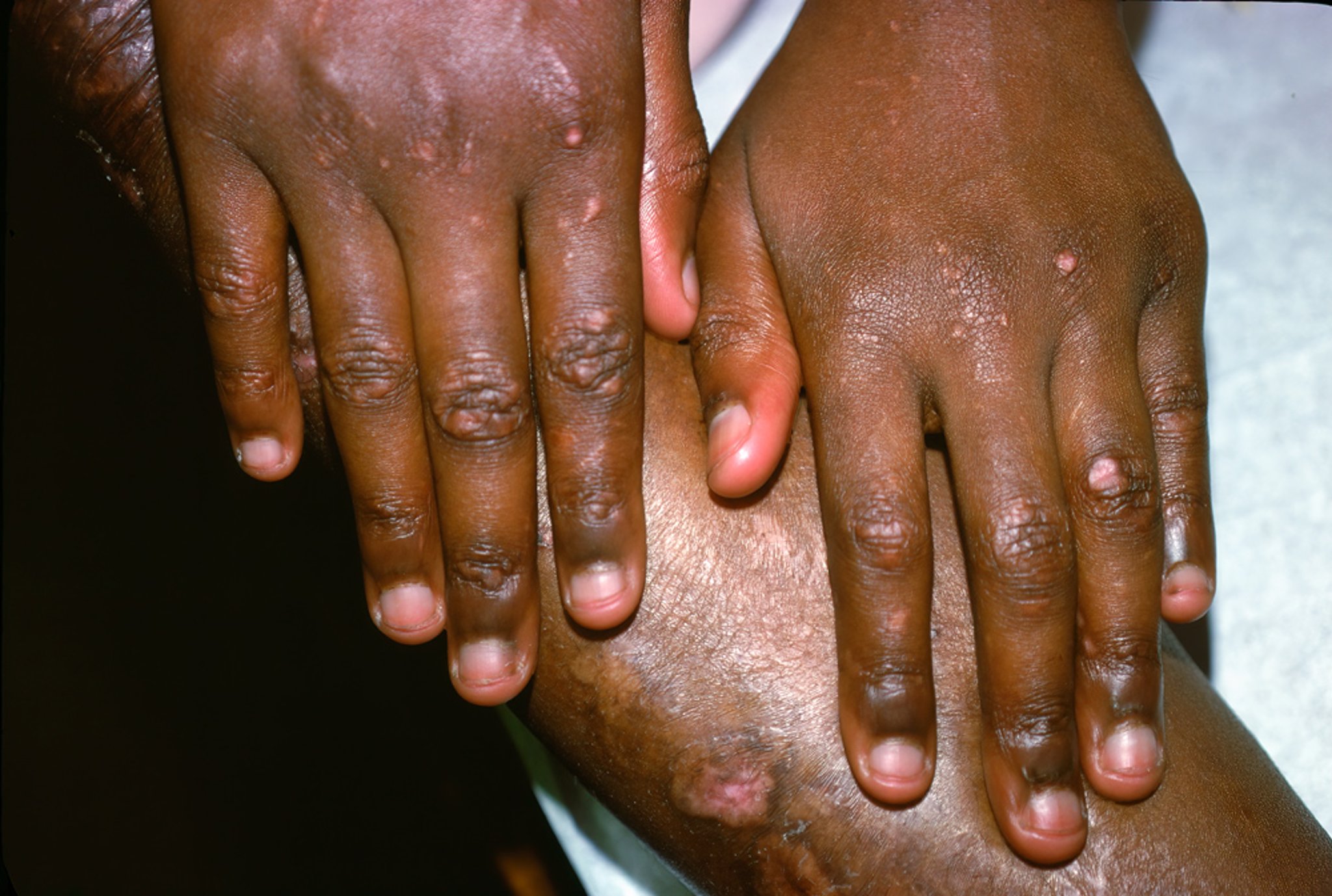
This photo shows hypopigmented scarring overlying bony prominences.
Photo courtesy of Karen McKoy, MD.
Kindler syndrome
Kindler syndrome has no subtypes.
Blistering particularly affects the dorsa of the hands and feet. Repeated episodes lead to progressive skin atrophy (thin, wrinkled skin), which can spread to other areas.
Photosensitivity, which may be mild, distinguishes Kindler syndrome from the other 3 major epidermolysis bullosa types.
Poikiloderma (constellation of skin atrophy, pigment changes, and telangiectasias) is common.
Mucocutaneous scarring leads to esophageal and genitourinary stenoses, ectropion, and pseudosyndactyly. Severe Kindler syndrome bullae heal with scarring because they occur in deeper levels.
Diagnosis of Epidermolysis Bullosa
Biopsy and immunofluorescence testing or transmission electron microscopy of skin
Gene mutation analysis
Epidermolysis bullosa is suspected based on clinical presentation. Family history may indicate the mode of inheritance and thus the possible type: autosomal dominant inheritance (epidermolysis bullosa simplex, dystrophic epidermolysis bullosa) or autosomal recessive inheritance (junctional epidermolysis bullosa, dystrophic epidermolysis bullosa, Kindler syndrome, rarely epidermolysis bullosa simplex).
Differential diagnosis includes common friction blisters and epidermolysis bullosa acquisita.
Diagnosis of specific types and subtypes is based on family history, biopsy and immunofluorescence testing or transmission electron microscopy of a freshly induced blister (not from the palms or soles), and gene mutation analysis of a blood sample.
Immunofluorescence testing or transmission electron microscopy determines the level of cleavage and bullae formation.
Genetic testing is usually done to confirm specific disease type and guide genetic counseling. (See table .)
Possible prenatal testing should be discussed with families that have a history of epidermolysis bullosa.
Treatment of Epidermolysis Bullosa
Avoidance of friction and trauma to skin and mucous membranes
Treatment to promote healing of blister sites
Prevention and treatment of complications
(See also Wound International's 2017 best practice guidelines for skin and wound care in epidermolysis bullosa.)
Treatment of epidermolysis bullosa is symptomatic; multidisciplinary specialization may likely be needed for both severe and chronic disease (1).
Any and all means of reducing skin friction are used. Neonates and infants are handled with exquisite gentleness. Soft clothing, padded furniture, and cool ambient temperatures help reduce blister formation.
A soft diet and stool softeners may be helpful if the gastrointestinal tract is affected.
Bullae should be properly incised and dressed, as instructed by a wound care specialist, to prevent further skin damage and infection. Minimally adherent dressings and tapes may be used; often, bandages covered with petroleum jelly are held in place with a light gauze wrap.
Medications are given as needed for pain, itching, and infection.
Surgical treatment may include gastrostomy tube placement, esophageal dilation, and correction of pseudosyndactyly and contractures.
Treatment reference
1. El Hachem M, Zambruno G, Bourdon-Lanoy E, et al: Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis 9:76, 2014. doi: 10.1186/1750-1172-9-76
Key Points
Epidermolysis bullosa is a group of inherited disorders causing bullous lesions of skin and mucous membranes.
Phenotypes vary widely in age of onset, severity, site of histologic defect, and inheritance pattern.
Diagnose using biopsy and immunofluorescence testing or transmission electron microscopy.
Treat supportively, often with multidisciplinary care.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Wound International: Best practice guidelines for skin and wound care in epidermolysis bullosa (2017)





