

Multiple endocrine neoplasia, type 2A (MEN 2A) is an autosomal dominant syndrome characterized by medullary carcinoma of the thyroid, pheochromocytoma, parathyroid hyperplasia or adenomas (causing hyperparathyroidism), and occasionally cutaneous lichen amyloidosis. Clinical features depend on the glandular elements affected. Familial medullary thyroid carcinoma is a distinct variant of MEN 2A. Diagnosis is confirmed by genetic testing. Hormonal and imaging tests help locate the tumors, which are removed surgically when possible.
(See also Overview of Multiple Endocrine Neoplasias.)
Mutations in the RET proto-oncogene on chromosome 10 have been identified in MEN 2A, MEN 2B, and familial medullary thyroid carcinoma. The RET protein is a receptor tyrosine kinase; MEN 2A and familial medullary thyroid carcinoma mutations result in activation of certain intracellular pathways.
Symptoms and Signs of MEN 2A
Clinical features depend on the type of tumor present (see table ).
Thyroid
Almost all patients have medullary thyroid carcinoma. The tumor usually develops during childhood and begins with thyroid parafollicular C-cell hyperplasia. Tumors are frequently multicentric.
Adrenal
Pheochromocytoma usually originates in the adrenal glands. Pheochromocytoma occurs in 40 to 50% of patients within a MEN 2A kindred. In contrast to sporadic pheochromocytoma, the familial variety within MEN 2A begins with adrenal medullary hyperplasia and is multicentric and bilateral in > 50% of cases. Extra-adrenal pheochromocytomas are rare. Pheochromocytomas are almost always benign, but some tend to recur locally and account for significant morbidity and mortality.
Pheochromocytomas that occur with MEN 2A (and MEN 2B) usually produce epinephrine disproportionately to norepinephrine, in contrast to sporadic cases.
Hypertensive crisis secondary to pheochromocytoma is a common manifestation. Hypertension in patients with MEN 2A and pheochromocytoma is more often paroxysmal than sustained, in contrast to the usual sporadic cases. Patients with pheochromocytomas may have paroxysmal palpitations, anxiety, headaches, or sweating; many are asymptomatic.
Parathyroid
Ten to 20% of patients have evidence of hyperparathyroidism (which may be long-standing), with hypercalcemia, nephrolithiasis, nephrocalcinosis, or renal failure. Hyperparathyroidism frequently involves multiple glands as either diffuse hyperplasia or multiple adenomas, and mild abnormalities in parathyroid function (not requiring surgical intervention) may also be present in MEN 2A.
Other manifestations
Cutaneous lichen amyloidosis, a pruritic, scaly, papular skin lesion, located in the interscapular region or on extensor surfaces, occurs in some MEN 2A-affected families. Hirschsprung disease is present in 2 to 5% of MEN 2A patients.

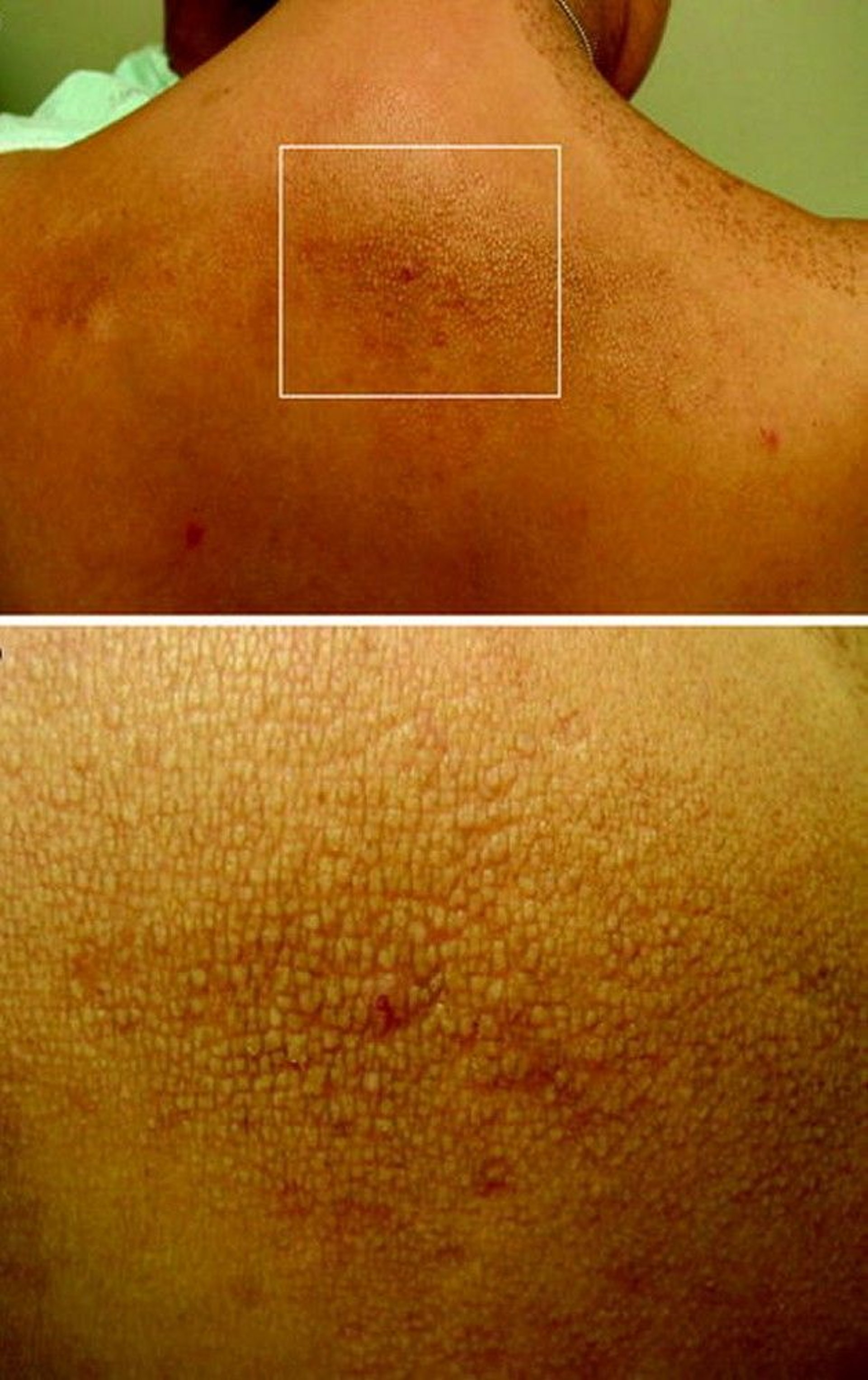
The top panel shows interscapular cutaneous lichen amyloidosis. The bottom panel shows a magnified view of the thickened, raised, scaly lesions typical of cutaneous lichen amyloidosis.
© Springer Science+Business Media

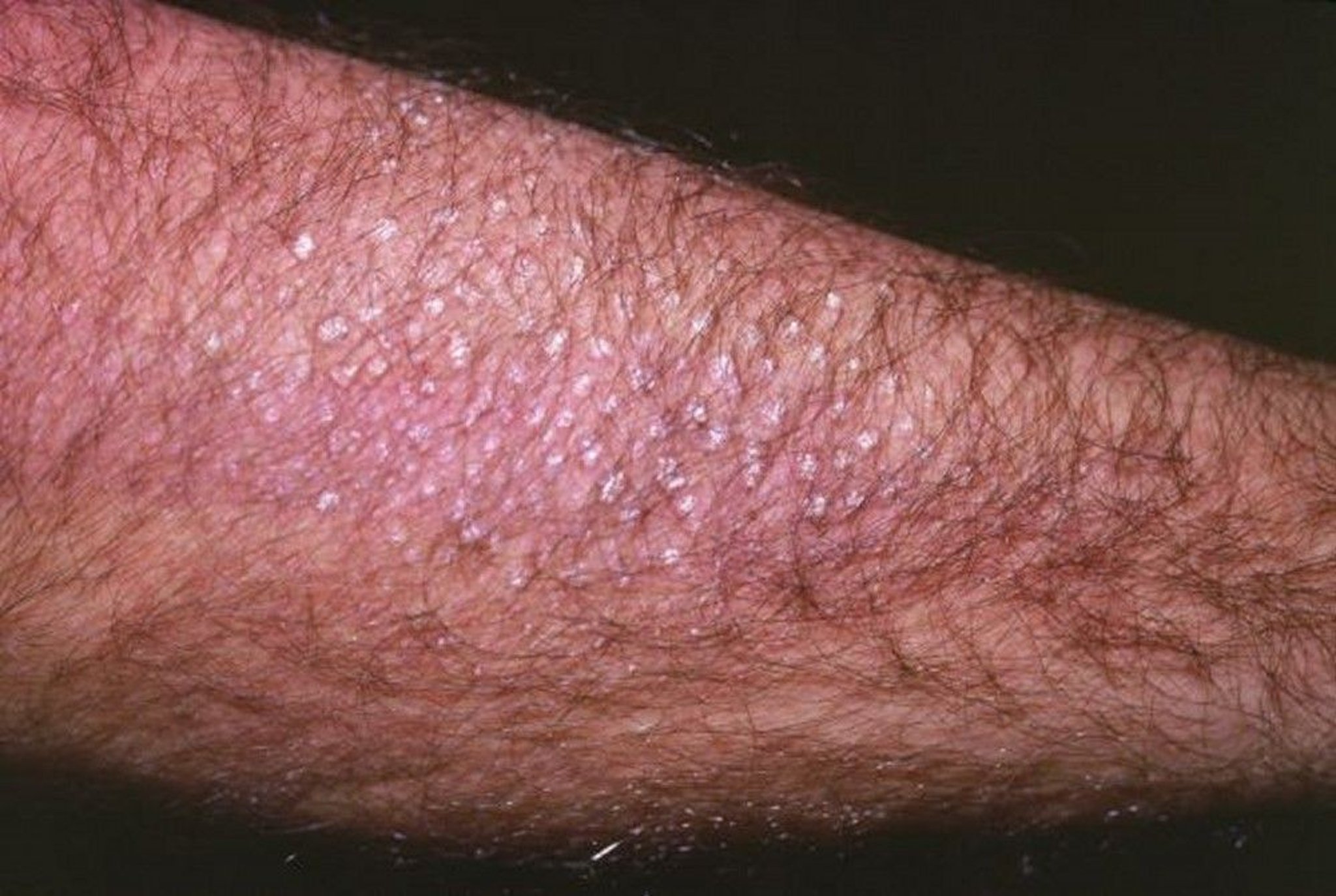
This image shows multiple thickened, raised, scaly lesions typical of cutaneous lichen amyloidosis.
Image courtesy of Karen McKoy, MD.
Diagnosis of MEN 2A
Serum calcitonin for medullary thyroid carcinoma
Serum calcium, 24-hour urine calcium, and parathyroid hormone for hyperparathyroidism
Plasma free metanephrines or urinary catecholamine and metanephrine levels for pheochromocytoma
Neck ultrasound, with additional imaging as needed
Localization of pheochromocytoma with MRI or CT
Genetic testing
Serum calcitonin is a good marker for the presence of medullary thyroid carcinoma and can also be used as a biomarker to follow disease response and/or progression.
Hyperparathyroidism is diagnosed by finding hypercalcemia, hypophosphatemia, and increased parathyroid hormone level.
Because pheochromocytoma may be asymptomatic, its exclusion may be difficult. The most sensitive tests are measurement of plasma free metanephrines or fractionated urinary catecholamines and metanephrines (particularly epinephrine).
Neck imaging for localization of masses within the thyroid and parathyroid glands should begin with ultrasound followed by CT, MRI, or radioisotope scanning as needed. CT or MRI is useful in localizing the pheochromocytoma or establishing the presence of bilateral lesions.
Many cases are identified during screening of family members of patients known to have MEN 2A. MEN 2A should also be suspected in patients with bilateral pheochromocytoma or at least 2 of its characteristic endocrine manifestations. The diagnosis can be confirmed with genetic testing. Although only 25% of medullary thyroid carcinoma cases are familial, genetic testing should be done in all patients with medullary thyroid carcinoma. RET mutations are found in virtually all patients with familial medullary thyroid cancer but are as high as approximately 7% in apparently sporadic cases (1).
Genetic screening
Genetic screening of family members of MEN 2A patients is the diagnostic test of choice; the availability of such testing has made biochemical screening for early medullary thyroid carcinoma largely obsolete. The specific RET mutation also predicts phenotypic characteristics such as aggressiveness of medullary thyroid carcinoma and presence of other endocrinopathies, so is important in clinical management; however, other factors, such as older age at onset and higher tumor stage at diagnosis, may be more predictive of disease aggressiveness (2).
Preimplantation genetic diagnosis and prenatal chorionic villus sampling or amniocentesis have been used for antenatal diagnosis.
In patients with affected family members, annual screening for hyperparathyroidism and pheochromocytoma should begin in adolescence (at 11 years of age for families with high-risk variants and at 16 years of age for those with moderate-risk variants) and continue indefinitely. Screening for hyperparathyroidism entails measurement of serum calcium levels. Screening for pheochromocytoma includes questions about symptoms, measurement of pulse rate and blood pressure, and laboratory testing.
Diagnosis references
1. Wells SA Jr, Asa SL, Dralle H, et al. American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25(6):567–610, 2015. doi:10.1089/thy.2014.0335
2. Voss RK, Feng L, Lee JE, et al. Medullary thyroid carcinoma in MEN2A: ATA moderate- or high-risk RET mutations do not predict disease aggressiveness. J Clin Endocrinol Metab 102(8):2807–2813, 2017. doi:10.1210/jc.2017-00317
Treatment of MEN 2A
Surgical excision of identified tumors
Prophylactic thyroidectomy
In patients presenting with pheochromocytoma and either medullary thyroid carcinoma or hyperparathyroidism, the pheochromocytoma should be removed first, even if asymptomatic because it greatly increases risk during other surgeries (1). Patients undergoing resection of a pheochromocytoma should receive adequate alpha blockade (typically using phenoxybenzamine, doxazosin, or prazosin) prior to surgery. Laparoscopic adrenalectomy, which has lower morbidity, is preferred to open laparotomy. Because bilateral pheochromocytomas are common, adrenal-sparing surgery may be appropriate in some patients (). Patients undergoing resection of a pheochromocytoma should receive adequate alpha blockade (typically using phenoxybenzamine, doxazosin, or prazosin) prior to surgery. Laparoscopic adrenalectomy, which has lower morbidity, is preferred to open laparotomy. Because bilateral pheochromocytomas are common, adrenal-sparing surgery may be appropriate in some patients (2).
Surgery for medullary thyroid carcinoma should include total thyroidectomy and central compartment lymph node dissection, with additional lymph node dissection if indicated based on preoperative imaging. Postsurgical assessment for residual or recurrent disease should include measurement of serum calcitonin and imaging with neck ultrasound and, when indicated, CT or MRI of neck and chest, bone scan, or positron emission tomography (PET) scan.
Once medullary thyroid carcinoma has metastasized, tyrosine kinase inhibitors, including selpercatinib, cabozantinib, vandetanib, and pralsetinib, can lengthen progression-free survival (Once medullary thyroid carcinoma has metastasized, tyrosine kinase inhibitors, including selpercatinib, cabozantinib, vandetanib, and pralsetinib, can lengthen progression-free survival (3). Clinical trials of other tyrosine kinase inhibitors and newer modalities such as chimeric antigen receptor (CAR)-T therapies continue to be developed (4). Cytotoxic chemotherapy and radiation therapy are largely ineffective in lengthening survival but may slow disease progression. Postoperative adjuvant external beam radiation should be considered in patients at high risk of local recurrence and those at risk for airway obstruction. Some studies have shown lengthened survival with immunotherapy (eg, tumor-derived vaccines, tumor cell transfectants) and radioimmunotherapy (eg, radioisotope-coupled monoclonal antibodies) (4).
Once genetic testing identifies a child as having a RET mutation, prophylactic thyroidectomy may be considered. Depending on the particular mutation, prophylactic thyroidectomy as early as the first months of life may be indicated. Medullary thyroid cancer can be cured or prevented by early thyroidectomy. There is a genotype-phenotype correlation for patients with RET mutations that may provide information about age of onset and clinical course of medullary thyroid cancer and thus impact the timing of surgery for an affected child (5, 6).
Psychological distress appears to be common and chronic in patients with MEN 2. Contributing factors include low amount of information on the disease, having children with the mutation, number of surgeries, and presence of comorbidities; psychological assessment to identify and treat affected individuals is recommended (7).
Treatment references
1. Wells SA Jr. Advances in the management of MEN2: From improved surgical and medical treatment to novel kinase inhibitors. Endocr Relat Cancer 25:T1–T13, 2018. doi:10.1530/ERC-17-0325
2. Castinetti F, Qi XP, Walz AL, et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: An international retrospective population-based study. Lancet Oncol 15(6):648–655, 2014. doi:10.1016/S1470-2045(14)70154-8
3. Zhang Y, Zheng WH, Zhou SH, et al. Molecular genetics, therapeutics and RET inhibitor resistance for medullary thyroid carcinoma and future perspectives. Cell Commun Signal 22(1):460, 2024. doi:10.1186/s12964-024-01837-x
4. Laganà M, Cremaschi V, Alberti A, Vodopivec Kuri DM, Cosentini D, Berruti A. The Evolving Treatment Landscape of Medullary Thyroid Cancer. Curr Treat Options Oncol 24(12):1815–1832, 2023. doi:10.1007/s11864-023-01145-5
5. Castinetti F, Eng C. Genotype/phenotype correlations in multiple endocrine neoplasia type 2. Endocr Relat Cancer 31(12):e240139, 2024. doi:10.1530/ERC-24-0139
6. Wells SA Jr, Asa SL, Dralle H, et al: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015;25(6):567-610. doi:10.1089/thy.2014.0335
7. Rodrigues KC, Toledo RA, Coutinho FL, et al. Assessment of depression, anxiety, quality of life, and coping in long-standing multiple endocrine neoplasia type 2 patients. Thyroid 27(5):693–706, 2017. doi:10.1089/thy.2016.0148
Key Points
Most patients with multiple endocrine neoplasia, type 2A have medullary thyroid carcinoma, typically beginning in childhood.
Other manifestations are those of hormone excess, particularly hypertension due to pheochromocytoma and hypercalcemia due to hyperparathyroidism.
Patients should have genetic testing for RET proto-oncogene mutations and clinical evaluation for other tumors of the syndrome.
Tumors are excised when possible, beginning with any pheochromocytoma.
Depending on the specific RET mutation, prophylactic thyroidectomy may be recommended; timing of the surgery may also be influenced by the specific mutation.
More Information
The following English-language resource may be useful. Please note that The Manual is not responsible for the content of this resource.
Saravana-Bawan B, Pasternak JD. Multiple endocrine neoplasia 2: an overview. Ther Adv Chronic Dis 13:20406223221079246, 2022. doi:10.1177/20406223221079246
Drug Information for the Topic





