

Gliomas are primary tumors that originate in brain parenchyma. Symptoms are diverse and vary by location, manifesting as focal neurologic deficits, encephalopathy, or seizures. Diagnosis is based primarily on MRI, including both standard T1- and T2-weighted imaging, preferably with gadolinium enhancement, followed by biopsy with molecular profiling. Treatment involves surgical excision, radiation therapy, and, for some tumors, chemotherapy. Excision rarely cures.
(See also Overview of Intracranial Tumors.)
Gliomas include
Astrocytomas
Oligodendrogliomas
Glioblastoma multiforme
Ependymomas
Many gliomas infiltrate brain tissue diffusely and irregularly.
Astrocytomas are the most common gliomas (see also Astrocytomas in children). They are classified histologically and, in some cases, based on the presence of specific genetic markers, according to the World Health Organization (WHO) classification (1).
In ascending order of malignancy, astrocytomas are classified as
Grade I: Pilocytic astrocytomas and subependymal giant cell astrocytomas (most common in tuberous sclerosis)
Grade II: Low-grade astrocytomas, including pleomorphic xanthoastrocytoma
Grade III: Anaplastic astrocytomas
Grade IV: Glioblastomas and diffuse midline gliomas
Pilocytic, other low-grade, or anaplastic astrocytomas tend to develop in younger patients. Anaplastic astrocytomas, in particular, can later evolve into glioblastomas (called secondary glioblastomas). Glioblastomas can also develop de novo (called primary glioblastomas), usually in middle-aged or older adults. Glioblastomas contain chromosomally heterogeneous cells. Both primary and secondary glioblastomas have distinct genetic characteristics, which can change as the tumors evolve. Secondary glioblastomas typically have mutations in the IDH1 or IDH2 genes.
Rarely, astrocytomas contain astrocytoma and oligodendroglioma cells. These tumors used to be designated oligoastrocytomas; however, that term is no longer used to refer to a single tumor type but rather to a mixed neoplasm.
Oligodendrogliomas (WHO grade II) are among the slowest-growing gliomas. They are most common in the forebrain, particularly the frontal lobes. Oligodendrogliomas are typically characterized by deletion of the p arm of chromosome 1 and the q arm of chromosome 19 (1p/19q codeletion). These deletions are diagnostic for oligodendroglial tumors, predict longer survival, and predict a better response to radiation therapy and chemotherapy. Like astrocytomas, oligodendrogliomas can evolve into more aggressive forms, such as anaplastic oligodendrogliomas (WHO grade III), which are managed accordingly.
Both astrocytomas and oligodendrogliomas may express mutations of the IDH1 or IDH2 genes that result in the abnormal production of 2-hydroxyglutarate; this metabolite can modify DNA methylation of normal neural and glial progenitor cells, causing them to produce neoplastic glioma cells. Patients with the IDH1/2 mutation tend to have a better prognosis than those with IDH1/2 wild-type tumors, partly because patients have a better response to alkylating chemotherapy such as temozolomide. Oligodendrogliomas tend to have the 1p/19q-codeletion and IDH1/2 mutation. Astrocytomas typically have the IDH1/2 mutation but not the 1p/19q codeletion; instead, they more typically express mutations or loss of the genes that result in the abnormal production of 2-hydroxyglutarate; this metabolite can modify DNA methylation of normal neural and glial progenitor cells, causing them to produce neoplastic glioma cells. Patients with the IDH1/2 mutation tend to have a better prognosis than those with IDH1/2 wild-type tumors, partly because patients have a better response to alkylating chemotherapy such as temozolomide. Oligodendrogliomas tend to have the 1p/19q-codeletion and IDH1/2 mutation. Astrocytomas typically have the IDH1/2 mutation but not the 1p/19q codeletion; instead, they more typically express mutations or loss of theATRX gene and mutations in pTP53 (2).
Diffuse midline gliomas are high-grade (WHO grade III to IV) astrocytic tumors that primarily affect children. These tumors include diffuse intrinsic pontine gliomas, which are aggressive and typically lethal tumors that infiltrate the brain stem with rostral extension into the hypothalamus and thalamus and that infiltrate the medulla and spinal cord inferiorly. Diffuse midline gliomas typically express the H3K27M mutation.
Children with neurofibromatosis type 1 are at an increased risk of developing diffuse midline gliomas.
Ependymomas occur primarily in children and young adults; they are uncommon after adolescence (see also Ependymomas in children). They are classified as
Grade I: Subependymoma
Grade II: Ependymoma
Grade III: Anaplastic ependymoma
Grade IV: Ependymoblastoma (which are rare and occur primarily in infants)
All ependymomas typically arise from the ventricular wall and hence may arise in the brain, brain stem, or spinal cord. As such, they are classified based on location: as supratentorial, posterior fossa, or spinal cord. Each of these categories includes 3 molecularly and histologically defined subsets, resulting in 9 types of phenotypically and molecularly distinct ependymomas, whose treatment and prognosis differ significantly from one another. Ependymomas of the fourth ventricle in particular can manifest with obstructive hydrocephalus and may therefore manifest earlier than other ependymomas (3).

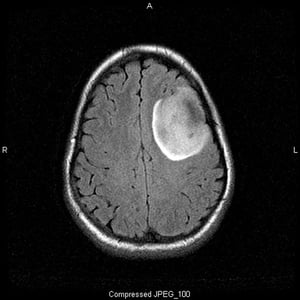
This T2-FLAIR (fluid-attenuating inversion recovery) MRI scan shows a white signal, which may indicate a mass or edema. The left frontal signal is highly demarcated, suggesting a mass. Use of contrast does not enhance it. It is a low-grade (grade II) oligodendroglioma.
This T2-FLAIR (fluid-attenuating inversion recovery) MRI scan shows a white signal, which may indicate a mass or edema.
Image courtesy of William R. Shapiro, MD.
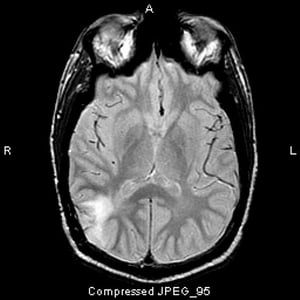
This T2-FLAIR MRI scan shows a white signal in the posterior temporal lobe. The signal is not enhanced by contrast. It is an anaplastic (grade III) astrocytoma.
This T2-FLAIR MRI scan shows a white signal in the posterior temporal lobe. The signal is not enhanced by contrast. It
Image courtesy of William R. Shapiro, MD.

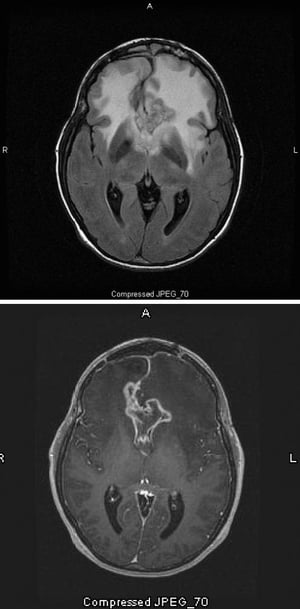
The T2-FLAIR MRI scan (top) shows a large, bilateral white (hyperdensity) signal around a glioblastoma, which is the highest grade and most malignant astrocytoma. This particular glioblastoma is called a butterfly glioma because the white signal around the tumor forms the wings of the butterfly; it is caused by cerebral edema. On the T1-weighted scan (bottom), contrast outlines the edge of the tumor (ring enhancement). The edema appears as a darkened area (hypointensity) on T1.
The T2-FLAIR MRI scan (top) shows a large, bilateral white (hyperdensity) signal around a glioblastoma, which is the hi
Images courtesy of William R. Shapiro, MD.
This T2-FLAIR (fluid-attenuating inversion recovery) MRI scan shows a white signal, which may indicate a mass or edema. The left frontal signal is highly demarcated, suggesting a mass. Use of contrast does not enhance it. It is a low-grade (grade II) oligodendroglioma.
This T2-FLAIR (fluid-attenuating inversion recovery) MRI scan shows a white signal, which may indicate a mass or edema.
Image courtesy of William R. Shapiro, MD.
This T2-FLAIR MRI scan shows a white signal in the posterior temporal lobe. The signal is not enhanced by contrast. It is an anaplastic (grade III) astrocytoma.
This T2-FLAIR MRI scan shows a white signal in the posterior temporal lobe. The signal is not enhanced by contrast. It
Image courtesy of William R. Shapiro, MD.
The T2-FLAIR MRI scan (top) shows a large, bilateral white (hyperdensity) signal around a glioblastoma, which is the highest grade and most malignant astrocytoma. This particular glioblastoma is called a butterfly glioma because the white signal around the tumor forms the wings of the butterfly; it is caused by cerebral edema. On the T1-weighted scan (bottom), contrast outlines the edge of the tumor (ring enhancement). The edema appears as a darkened area (hypointensity) on T1.
The T2-FLAIR MRI scan (top) shows a large, bilateral white (hyperdensity) signal around a glioblastoma, which is the hi
Images courtesy of William R. Shapiro, MD.
General references
1. Louis DN, Perry A, Reifenberger G, et al: The 2021 World Health Organization classification of tumors of the central nervous system: A summary. Neuro Oncol 23 (8):1231–1251, 2021. doi: 10.1093/neuonc/noab106
2. Reifenberger G, Wirsing H, Knobbe-Thomsen CB, Weller M: Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 14 (7):434-452 2017. doi: 10.1038/nrclinonc.2016.204
3. Pajtler K, Mack S, Ramaswamy V, et al: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Neuropathologica 133:5–12, 2017. doi: 10.1007/s00401-016-1643-0
Diagnosis of Gliomas
T1- and T2-weighted MRI
Biopsy
Diagnosis of gliomas is based primarily on MRI, including both standard T1- and T2-weighted imaging, preferably with gadolinium enhancement, followed by biopsy with histopathology and molecular profiling. Histopathology includes classical pathologic analysis of cellular morphology, immunohistochemical stains (eg, for IDH1/2 mutations), and in situ hybridization (eg, for EGFR mutations).
Selected oncogene panels, targeted or whole-exome sequencing, RNA sequencing, and/or methylguanine-DNA methyl-transferase (MGMT) methylation analysis may be done. Methylation of the MGMT gene promoter is a prognostic factor in patients with glioblastoma multiforme; it predicts a good response to postoperative temozolamide and increased survival (1).
Diagnosis reference
1. Stupp R, Taillibert S, Kanner A, et al: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. : Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial.JAMA 318 (23):2306–2316, 2017. doi: 10.1001/jama.2017.18718
Treatment of Gliomas
Surgical excision
Radiation therapy
Chemotherapy for some types
Anaplastic astrocytomas and glioblastomas
Treatment involves surgery, radiation therapy, and chemotherapy to reduce tumor mass. Safely excising as much tumor as possible (without unduly damaging areas of eloquent brain [areas that subserve functions such as speech and motor function]) prolongs survival and improves neurologic function.
After surgery, patients receive a full tumor dose of radiation therapy (60 Gy over 6 weeks); ideally, conformal radiation therapy, which targets the tumor and spares normal brain tissue, is used.
For glioblastomas, chemotherapy with temozolomide is now routinely given with radiation therapy. For glioblastomas, chemotherapy with temozolomide is now routinely given with radiation therapy.
During treatment with temozolomide, trimethoprim/sulfamethoxazole 800 mg/160 mg is given 3 times a week to prevent During treatment with temozolomide, trimethoprim/sulfamethoxazole 800 mg/160 mg is given 3 times a week to preventPneumocystis jirovecii pneumonia.
Patients receiving chemotherapy require a complete blood count (CBC) at varying intervals.
Implantation of chemotherapy wafers during surgical resection may be appropriate for some patients.
Use of tumor-treating fields plus adjuvant temozolomide may be appropriate for some patients. Tumor-treating fields interfere with glioblastoma mitosis and organelle assembly by delivering alternating electric fields to the scalp. Tumor-treating fields appear to improve survival in patients with glioblastoma (Use of tumor-treating fields plus adjuvant temozolomide may be appropriate for some patients. Tumor-treating fields interfere with glioblastoma mitosis and organelle assembly by delivering alternating electric fields to the scalp. Tumor-treating fields appear to improve survival in patients with glioblastoma (1, 2).
Investigational therapies (eg, stereotactic radiosurgery, new chemotherapeutic agents, gene or immune therapy) should also be considered. Clinical trials of numerous immune checkpoint inhibitors, in particular of CTLA4- and PD1-dependent immune modulation, are underway.
After conventional multimodal treatment, the survival rate for patients with glioblastomas is about 50% at 1 year, 25% at 2 years, and 10 to 15% at 5 years (3). Prognosis is better in the following cases:
Patients are < 45 years (4).
Histology is that of anaplastic astrocytoma or a lower-grade tumor (rather than glioblastoma) (4).
Initial excision improves neurologic function and leaves minimal or no residual tumor.
Tumors have the IDH1 mutation (5).
MGMT (methylguanine-methyltransferase) promoter methylation is present (2).
With standard treatment, the median survival time is about 26 months for patients with anaplastic astrocytoma and about 13 months for patients with glioblastoma (6). A wide range of clinical trials, in particular those including a number of investigational immunotherapy and immune checkpoint inhibitors, are being done. Ongoing and recently completed trials are referenced in clinicaltrials.gov.
Low-grade astrocytomas and low-grade oligodendrogliomas
Maximal safe surgical resection is indicated for low-grade astrocytomas and oligodendrogliomas. After complete resection in patients < 40, observation may be considered. For other patients, radiation therapy plus adjuvant chemotherapy prolongs survival (7).
Median survival ranges from 1 to 2 years in high-risk patients (without the IDH1 mutation, with incomplete resection) to > 10 years in those with favorable prognostic factors (8). In high-risk patients, malignancy is likely to progress further.
Diffuse midline gliomas
In patients with diffuse midline gliomas, radiation therapy may be used to slow disease progression, although its use is largely palliative because survival is typically less than a year. These tumors typically express the H3K27M mutation in a histone protein; epigenetic strategies that modify DNA and DNA-associated proteins in the cell to change gene expression are being studied (9). Patients with the H3K27M mutation have a poor prognosis regardless of the histologic grade of the tumor.
Ependymomas
Biopsy with genetic analysis, cerebrospinal fluid sampling, and craniospinal imaging should be used to stage ependymomas and to assess their spread in the central nervous system.
Treatment of ependymomas includes maximal safe surgical resection for unifocal disease or symptomatic tumors. For higher-grade tumors, radiation therapy can be directed locally or include the entire craniospinal axis depending on how far the tumor has spread. The role of chemotherapy is not well defined.
Prognosis varies with tumor location, genetics and stage (10). In general, with treatment, overall 5-year survival rate is approximately ≥ 50% (11); however, for patients with no residual tumor, the 5-year survival rate higher.
Treatment references
1. Ballo MT, Conlon P, Lavy-Shahaf G, et al: Association of tumor treating fields (TTFields) therapy with survival in newly diagnosed glioblastoma: A systematic review and meta-analysis. J Neurooncol 164(1):1-9, 2023. doi: 10.1007/s11060-023-04348-w
2. Stupp R, Taillibert S, Kanner A, et al: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. : Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial.JAMA 318 (23):2306–2316, 2017. doi: 10.1001/jama.2017.18718
3. Brown NF, Ottaviani D, Tazare J, et al: Survival outcomes and prognostic factors in glioblastoma. Cancers (Basel) 14(13):3161, 2022. doi: 10.3390/cancers14133161.
4. Liang J, Lv X, Lu C, et al: Prognostic factors of patients with gliomas – An analysis on 335 patients with glioblastoma and other forms of gliomas. BMC Cancer 20:35, 2020. https://doi.org/10.1186/s12885-019-6511-6Download citation
5. Cancer Genome Atlas Research Network; Brat DJ, Verhaak RG, Aldape KD, M et al: Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372(26):2481-2498, 2015. doi: 10.1056/NEJMoa1402121.
6. Noiphithak R, Veerasarn K: Clinical predictors for survival and treatment outcome of high-grade glioma in Prasat Neurological Institute. Asian J Neurosurg 12(1):28-33, 2017. doi: 10.4103/1793-5482.148791
7. Buckner JC, Shaw EG, Pugh SL, et al: Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. : Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma.N Engl J Med 374(14):1344-1355, 2016. doi: 10.1056/NEJMoa1500925
8. Choi J, Kim SH, et al: Extent of resection and molecular pathologic subtype are potent prognostic factors of adult WHO grade II glioma. Sci Rep 10(1):2086, 2020. doi: 10.1038/s41598-020-59089-x
9. Miklja Z, Pasternak A, Stallard S, et al: Molecular profiling and targeted therapy in pediatric gliomas: Review and consensus recommendations. Neuro Oncol 21:968–980, 2019.
10. Pajtler K, Mack S, Ramaswamy V, et al: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Neuropathologica 133:5–12, 2017. doi: 10.1007/s00401-016-1643-0
11. McGuire CS, Sainani KL, Fisher PG: Both location and age predict survival in ependymoma: A SEER study. Pediatric Blood Cancer 52(1):65-69, 2008. Wiley Online Library. https://doi.org/10.1002/pbc.21806
Key Points
Gliomas are primary tumors that originate in brain parenchyma; they include astrocytomas, oligodendrogliomas, and ependymomas.
Gliomas vary in location, degree of malignancy, treatment, and prognosis.
For most gliomas, surgically excise as much tumor as possible (gross total resection) without unduly damaging areas of eloquent brain, then follow with radiation therapy and/or chemotherapy.
Drug Information for the Topic





