




Craniosynostosis is premature fusion of one or more calvarial sutures.
(See also Introduction to Congenital Craniofacial and Musculoskeletal Disorders and Overview of Congenital Craniofacial Abnormalities.)
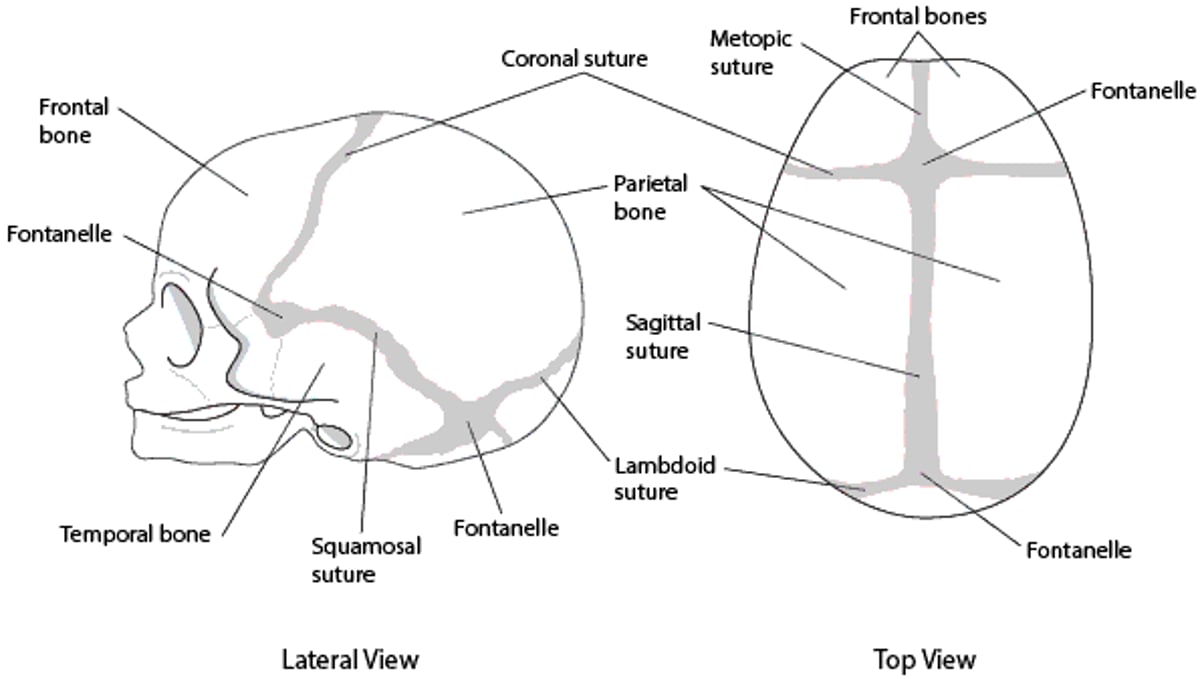
Premature fusion of sutures causes a characteristic skull deformity due to decreased growth in a direction perpendicular to the closed suture. It occurs in 1 of 2500 live births. There are several types, depending on which suture is fused.
Skull sutures

A clinical geneticist should evaluate affected patients even in cases of apparent isolated congenital anomaly.
Chromosomal microarray analysis, specific gene tests, or broader gene panel tests should be considered in the evaluation of patients with congenital craniofacial abnormalities. If the results of these tests are nondiagnostic, whole exome sequencing analysis is recommended.
Sagittal craniosynostosis
Sagittal craniosynostosis is the most common type and causes a narrow and long skull (dolichocephaly). Most cases are isolated and sporadic, with recurrence risk of transmission to future offspring < 3%. Learning disability may be present in up to 40 to 50% of patients. Several genes have been implicated in sagittal craniosynostosis, but chromosomal microarray analysis is not typically necessary unless developmental delays or other congenital anomalies are present.

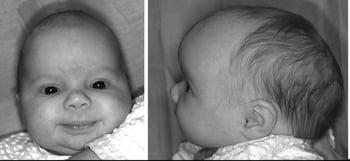
Coronal craniosynostosis
Coronal craniosynostosis is the second most common type and can be bilateral, causing a short and broad skull (brachycephaly), or unilateral, causing a diagonal skull deformity (plagiocephaly). True plagiocephaly (ie, caused by craniosynostosis) often results in asymmetric orbits and is to be differentiated from positional plagiocephaly, which is due to torticollis or positioning the infant predominantly on one side and does not result in asymmetric orbits. In positional plagiocephaly, the back of the skull is flattened on one side, there is frontal bossing on the same side, and the ear on the flattened side may be pushed forward, but the orbits remain symmetrical.
About 25% of coronal craniosynostosis cases are syndromic and due to single-gene mutations or chromosomal defects. Mutations in several genes have been identified in patients with isolated nonsyndromic coronal craniosynostosis. Specifically, 32% of patients with bilateral coronal craniosynostosis and 10% of patients with unilateral coronal craniosynostosis have mutations in the TCF12 gene (1). A specific gene panel test is currently recommended even in sporadic cases.
Coronal craniosynostosis is commonly associated with facial and extracranial anomalies within the context of Crouzon, Muenke, Pfeiffer, Saethre-Chotzen, Carpenter, or Apert syndromes.


Coronal craniosynostosis reference
1. Sharma VP, Fenwick AL, Brockop MS, et al: Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet 45(3):304–307, 2013. doi: 10.1038/ng.2531. Epub 2013 Jan 27. Clarification and additional information. Nat Genet 45(10):1261, 2013.







