

Erythropoietic protoporphyria (EPP) is due to an inherited deficiency in the activity of the enzyme ferrochelatase. X-linked protoporphyria (XLPP) is due to an inherited increase in the activity of delta-aminolevulinic acid synthase-2. Enzymes that cause both erythropoietic protoporphyria and X-linked protoporphyria are in the heme biosynthetic pathway (see table ). Erythropoietic protoporphyria and X-linked protoporphyria are nearly identical clinically. They typically manifest in infancy with itching or burning skin pain after even short exposure to sunlight. Gallstones are common later in life, and chronic liver disease occurs in about 10%. Diagnosis is based on symptoms and increased levels of protoporphyrin in red blood cells and plasma. Prevention is by avoidance of triggers (eg, sunlight, alcohol, fasting). Available therapies can decrease photosensitivity. Acute skin symptoms can be treated by cold baths or wet towels, analgesics, and topical and/or oral corticosteroids, although objective evidence for benefit is weak. Patients with liver failure may need liver transplantation, but liver transplantation is not curative because the predominant source of excess protoporphyrin production is the bone marrow. Bone marrow transplantation is curative, albeit high risk and rarely indicated.
(See also Overview of Porphyrias and Overview of Cutaneous Porphyrias.)
Because X-linked protoporphyria is so similar to erythropoietic protoporphyria, it is sometimes regarded as a variant of erythropoietic protoporphyria.
Etiology
Erythropoietic protoporphyria, which comprises about 90% of erythropoietic protoporphyria phenotypic presentations, results from inherited deficiency of the enzyme ferrochelatase. The inheritance pattern is autosomal recessive; thus, clinical manifestations occur only in people with 2 defective FECH alleles, or more commonly, one defective and one low-expressing wild-type allele (1).
X-linked protoporphyria, which comprises the remaining 10% of cases, results from gain-of-function mutations that increase the activity of erythroid-specific delta-aminolevulinate synthase (ALAS 2) in the bone marrow; inheritance is X-linked. The phenotype of heterozygous females can vary from asymptomatic to that of affected males (1).
Prevalence of erythropoietic protoporphyria phenotype is about 1/75,000. However, a study from the UK Biobank found that the prevalence of erythropoietic protoporphyria was about 1/17,000 (2). The authors attributed this higher prevalence to under diagnosis. Protoporphyrin accumulates in bone marrow and red blood cells, enters the plasma, and is deposited in the skin or excreted by the liver into bile. About 10% of patients develop chronic liver disease; a few of these patients develop cirrhosis, which may progress to liver failure. A more common complication is pigment gallstones due to heavy protoporphyrin excretion.
Etiology references
1. Balwani M, Doheny D, Bishop DF, et al: Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med 19(1):26–35, 2013. doi:10.2119/molmed.2012.00340
2. Dickey AK, Quick C, Ducamp S, et al: Evidence in the UK Biobank for the underdiagnosis of erythropoietic protoporphyria. Genet Med 23(1):140–148, 2021. doi:10.1038/s41436-020-00951-8
Symptoms and Signs
Symptom severity in erythropoietic protoporphyria and X-linked protoporphyria varies greatly, even among patients within a single family. One study suggests that higher levels of erythrocyte protoporphyrin are a major determinant of disease severity and risk of liver dysfunction in patients with erythropoietic protoporphyria or X-linked protoporphyria (1).
Most patients develop symptoms in early childhood. Brief exposure to sunlight can cause severe pain, burning, erythema, and edema of the exposed skin. Usually, an infant or young child cries for hours after even short exposure to sun. Patients are often erroneously said to suffer from "sun allergy." Sometimes skin swelling and erythema may be subtle or absent, and erythropoietic protoporphyria and X-linked protoporphyria may go undiagnosed longer than any other of the porphyrias.

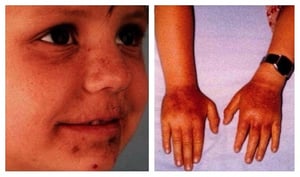
The left photo shows erythema, hyperpigmentation and crusting on the face of a young girl with erythropoietic protoporphyria after sun exposure. The photo on the right shows erythema and edema of the hands and wrists of a young boy with erythropoietic protoporphyria after sun exposure.
The left photo shows erythema, hyperpigmentation and crusting on the face of a young girl with erythropoietic protoporp
© Springer Science+Business Media

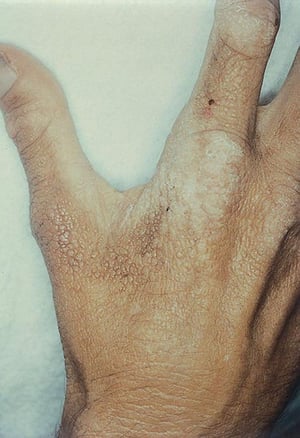
In erythropoietic protoporphyria, chronic skin changes include thickening and lichenification of skin over the dorsal aspect of the hand.
In erythropoietic protoporphyria, chronic skin changes include thickening and lichenification of skin over the dorsal a
By permission of the publisher. From Bloomer J, Risheg H. In Gastroenterology and Hepatology: Liver. Edited by M Feldman (series editor) and WC Maddrey. Philadelphia, Current Medicine, 2004.

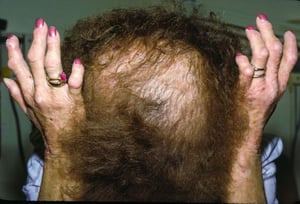
This image shows the chronic skin changes (rough, thickened, and leathery skin [lichenification]) that may develop, especially over the knuckles and scalp, in erythropoietic protoporphyria.
This image shows the chronic skin changes (rough, thickened, and leathery skin [lichenification]) that may develop, esp
Image courtesy of Karen McKoy, MD.

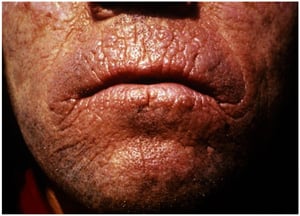
This photo shows linear perioral furrows (carp mouth) in a person with erythropoietic protoporphyria.
This photo shows linear perioral furrows (carp mouth) in a person with erythropoietic protoporphyria.
© Springer Science+Business Media
The left photo shows erythema, hyperpigmentation and crusting on the face of a young girl with erythropoietic protoporphyria after sun exposure. The photo on the right shows erythema and edema of the hands and wrists of a young boy with erythropoietic protoporphyria after sun exposure.
The left photo shows erythema, hyperpigmentation and crusting on the face of a young girl with erythropoietic protoporp
© Springer Science+Business Media
In erythropoietic protoporphyria, chronic skin changes include thickening and lichenification of skin over the dorsal aspect of the hand.
In erythropoietic protoporphyria, chronic skin changes include thickening and lichenification of skin over the dorsal a
By permission of the publisher. From Bloomer J, Risheg H. In Gastroenterology and Hepatology: Liver. Edited by M Feldman (series editor) and WC Maddrey. Philadelphia, Current Medicine, 2004.
This image shows the chronic skin changes (rough, thickened, and leathery skin [lichenification]) that may develop, especially over the knuckles and scalp, in erythropoietic protoporphyria.
This image shows the chronic skin changes (rough, thickened, and leathery skin [lichenification]) that may develop, esp
Image courtesy of Karen McKoy, MD.
This photo shows linear perioral furrows (carp mouth) in a person with erythropoietic protoporphyria.
This photo shows linear perioral furrows (carp mouth) in a person with erythropoietic protoporphyria.
© Springer Science+Business Media
Crusting may develop around the lips and on the back of the hands after prolonged sun exposure. However, blistering and scarring, as are typical in porphyria cutanea tarda, variegate porphyria, and congenital erythropoietic porphyria (see table ), do not occur.
If skin protection is chronically neglected, rough, thickened, and leathery skin (lichenification) may develop, especially over the knuckles. Linear perioral furrows (carp mouth) may develop. Patients with X-linked protoporphyria tend to have more severe photosensitivity and liver disease than those with erythropoietic protoporphyria.
If unrecognized, erythropoietic protoporphyria and X-linked protoporphyria may cause psychosocial problems because children inexplicably refuse to go outdoors. The fear or anticipation of pain may be so distressing that children become nervous, tense, aggressive, or even develop feelings of detachment from the surroundings or suicidal thoughts.
Symptoms and signs reference
1. Balwani M, Naik H, Anderson KE, et al: Clinical, biochemical, and genetic characterization of North American patients with erythropoietic protoporphyria and X-linked protoporphyria. JAMA Dermatol 153(8):789–796, 2017. doi:10.1001/jamadermatol.2017.1557
Diagnosis
Red blood cell and plasma total and metal-free protoporphyrin measurement
Genetic testing for FECH or ALAS 2 gene mutations
Erythropoietic protoporphyria or X-linked protoporphyria should be suspected in children and adults with painful cutaneous photosensitivity who experience no blisters or scarring. Gallstones in children should prompt testing for erythropoietic protoporphyria and X-linked protoporphyria. Family history is usually negative.
The diagnosis is confirmed by finding increased red blood cell and plasma protoporphyrin levels. The peak plasma fluorescence occurs at 634 nm, following excitation at 410 nm. Protoporphyrin levels in the blood vary significantly among patients with X-linked protoporphyria but are generally higher than in those with erythropoietic protoporphyria.
Red blood cell protoporphyrin should also be fractionated to determine the proportions of metal-free and zinc protoporphyrin. In erythropoietic protoporphyria, the proportion of red blood cell protoporphyrin that is metal-free is almost always > 85%. The presence of > 15% zinc protoporphyrin suggests X-linked protoporphyria.
If measured, plasma coproporphyrin and urinary porphyrin levels are normal. Stool protoporphyrin may be elevated, but coproporphyrin level is normal.
Potential carriers among relatives can be identified by finding increased red blood cell protoporphyrin levels and by genetic testing if a mutation has been identified in the index case.
Treatment
Avoidance of sun exposure through use of protective clothing and opaque sunscreens
Afamelanotide or dersimelagon for prevention of phototoxic events and amelioration of phototoxic reactionsAfamelanotide or dersimelagon for prevention of phototoxic events and amelioration of phototoxic reactions
Symptomatic treatment for skin burning with cold compresses, nonsteroidal anti-inflammatory drugs (NSAIDs), and topical and/or oral corticosteroids
Management of hepatobiliary complications
Treatment for erythropoietic protoporphyria and X-linked protoporphyria is similar. Patients with erythropoietic protoporphyria or X-linked protoporphyria should avoid sun exposure; protective clothing, hats, and light-opaque titanium dioxide or zinc oxide containing sunscreens should be used. Treatment for erythropoietic protoporphyria and X-linked protoporphyria is similar. Patients with erythropoietic protoporphyria or X-linked protoporphyria should avoid sun exposure; protective clothing, hats, and light-opaque titanium dioxide or zinc oxide containing sunscreens should be used.
Afamelanotide, a long-acting congener of melanocyte stimulating hormone which leads to increased production of eumelanin, can be administered once every 60 days as a subcutaneous implant, decreases photosensitivity, allows longer exposure to light, and improves quality of life (Afamelanotide, a long-acting congener of melanocyte stimulating hormone which leads to increased production of eumelanin, can be administered once every 60 days as a subcutaneous implant, decreases photosensitivity, allows longer exposure to light, and improves quality of life (1).
Dersimelagon, an orally administered selective melanocortin 1 receptor agonist increases production of eumelanin with resultant photoprotective effects. In a phase 2 trial, dersimelagon increased the duration of symptom-free sunlight exposure in erythropoietic protoporphyria and X-linked protoporphyria (2).
Oral beta-carotene, an antioxidant, reduces photosensitivity. However, patient adherence with beta-carotene is often poor because it is not very effective in controlling symptoms and also causes orange skin pigmentation, which is less noticeable and less severe in patients with dark skin; thus, beta-carotene is often tried but usually not continued. Beta-carotene dose depends on patient’s age.
Medications that trigger acute porphyrias need not be avoided.
In some patients, acute skin symptoms can be alleviated by cold baths or wet towels, analgesics, and topical and/or oral corticosteroids. However, many patients do not find such measures very effective. Symptoms can take up to a week to resolve.
If these measures are ineffective (eg, patients have increasing photosensitivity, rising porphyrin levels, progressive jaundice), giving hematin and/or red blood cell hypertransfusion (ie, to above-normal hemoglobin levels) may reduce protoporphyrin overproduction. Plasmapheresis is also effective acutely to decrease high concentrations of protoporphyrin in plasma; however, the duration of the effect is often short-lived.
Administration of bile acids may facilitate biliary excretion of protoporphyrin.
Oral cholestyramine or charcoal have been used to interrupt the enterohepatic circulation of protoporphyrin and increase fecal excretion. Oral cholestyramine or charcoal have been used to interrupt the enterohepatic circulation of protoporphyrin and increase fecal excretion.
Oral vitamin E, 800 units per day, is also used with the hope that it will exert an antioxidant and hepatoprotective effect, although objective evidence of benefit is scant.Oral vitamin E, 800 units per day, is also used with the hope that it will exert an antioxidant and hepatoprotective effect, although objective evidence of benefit is scant.
Patients with erythropoietic protoporphyria or X-linked protoporphyria, especially women, are often anemic and iron-deficient for reasons that are not well understood. It is not due to abnormalities in hepcidin, the hormone produced in the liver that decreases iron absorption and release from storage (3). Iron replacement usually leads to decreased levels of protoporphyrin and to decreased severity of photosensitivity in X-linked protoporphyria but to increased levels of protoporphyrin in erythropoietic protoporphyria (4). Patients with erythropoietic protoporphyria should not be given iron replacement by IV, because of risks of triggering acute worsening of hepatic disease. If needed, oral iron, given once every day or every other day, with vitamin C, is the preferred treatment.
Patients who develop decompensated end-stage liver disease require liver transplantation. As with acute intermittent porphyria, patients with erythropoietic protoporphyria are not eligible for standardized Model for End-Stage Liver Disease (MELD)-exception points. However, liver transplantation does not correct the underlying metabolic defect, and erythropoietic protoporphyria hepatopathy often develops in the transplanted liver.
Hematopoietic stem cell transplantation is curative for erythropoietic protoporphyria but is not routinely done because the risk typically outweighs the benefits. The strategy of hematopoietic stem cell transplantation after liver transplantation cures erythropoietic protoporphyria and prevents recurrent erythropoietic protoporphyria from damaging the allograft, but the optimal timing of this strategy has not been established. Patients should be protected from operating room lights during liver transplantation or other prolonged surgery to avoid serious phototoxic injury to internal organs. Light sources should be covered with commercially available filters that block wavelengths ~380 to 420 nm (5). Endoscopy, laparoscopy, and brief (< 1.5 hour) abdominal surgery do not usually cause phototoxic damage.
Pearls & Pitfalls
|
Regular physician-patient consultations that provide information, discussion, and opportunities for genetic counseling together with physical checkups are important. Liver function and red blood cell and plasma protoporphyrin levels should be checked annually.
Patients with abnormal liver function test results should be evaluated by a hepatologist; a liver biopsy may be needed to stage the degree of fibrosis. Patients with known chronic liver disease should undergo screening liver ultrasonography, CT, or MRI every 6 months to check for hepatocellular carcinoma.
Vitamin D levels should be checked because deficiency is common (patients tend to avoid sun exposure); supplements are given if levels are low.
All patients with erythropoietic protoporphyria and X-linked protoporphyria should receive hepatitis A vaccine and hepatitis B vaccine and be advised to avoid alcohol.
Any chemicals, medications, or conditions cause cholestasis may lead to worsening of erythropoietic protoporphyria or X-linked protoporphyria and should be avoided.
Treatment references
1. Langendonk JG, Balwani M, Anderson KE, et al: Afamelanotide for erythropoietic protoporphyria. N Engl J Med 373:48–59, 2015. doi: 10.1056/NEJMoa1411481
2. Balwani M, Bonkovsky HL, Levy C, et al: Dersimelagon in Erythropoietic Protoporphyrias. N Engl J Med 388(15):1376–1385, 2023. doi:10.1056/NEJMoa2208754
3. Bossi K, Lee J, Schmeltzer P, et al: Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur J Clin Invest 45(10):1032–1041, 2015. doi:10.1111/eci.12503
4. Barman-Aksözen J, Minder EI, Schubiger C, Biolcati G, Schneider-Yin X: In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol Dis 54(1):71–77, 2015. doi:10.1016/j.bcmd.2014.07.017
5. Hanaki T, Noda T, Eguchi H, et al. Successful Liver Transplantation for Liver Failure With Erythropoietic Protoporphyria by Covering the Operating Theater Lights With Polyimide Film: A Case Report. Transplant Proc 52(2):625–629, 2020. doi:10.1016/j.transproceed.2019.12.004
Key Points
Erythropoietic protoporphyria causes severe pain, burning, erythema, and edema of exposed skin even after only brief exposure to sunlight; symptoms are not brought on by medications that trigger other porphyrias.
Cirrhosis develops in about 10% of patients, sometimes progressing to liver failure.
Measure red blood cell and plasma protoporphyrin levels.
Prevent photosensitivity symptoms by avoiding sun exposure and sometimes using medication (eg, beta-carotene, afamelanotide).Prevent photosensitivity symptoms by avoiding sun exposure and sometimes using medication (eg, beta-carotene, afamelanotide).
Hematin and/or red blood cell hypertransfusion may reduce protoporphyrin overproduction.
X-linked protoporphyria is clinically similar to erythropoietic protoporphyria, but photosensitivity and liver disease are more severe than in erythropoietic protoporphyria.
A useful clue for X-linked protoporphyria is a high proportion of red blood cell protoporphyrin that is zinc protoporphyrin.
Management of X-linked protoporphyria is similar to that of erythropoietic protoporphyria.
More Information
The following English-language resources may be useful. Please note that The Manual is not responsible for the content of these resources.
American Porphyria Foundation: Aims to educate and support patients and families affected by porphyrias and to support research into treatment and prevention of porphyrias
American Porphyria Foundation: Safe/Unsafe Drug Database: Provides a list of medications available in the United States to assist physicians in prescribing for patients with porphyrias
European Porphyria Network: Promotes clinical research about porphyrias
The Drug Database for Acute Porphyrias: Provides an up-to-date list of medications available in Europe to assist physicians in prescribing for patients with porphyrias
United Porphyrias Association: Provides education and support to patients and their families; provides reliable information to health-care providers; fosters and supports clinical research to improve diagnosis and management of the porphyrias
Levy C, Dickey AK, Wang B, et al. Evidence-based consensus guidelines for the diagnosis and management of protoporphyria-related liver dysfunction in erythropoietic protoporphyria and X-linked protoporphyria. Hepatology 2024;79(3):731-743. doi:10.1097/HEP.0000000000000546
Balwani M, Naik H, Anderson KE, et al. Clinical, biochemical, and genetic characterization of North American patients with erythropoietic protoporphyria and X-linked protoporphyria. JAMA Dermatol 153(8):789–796, 2017. doi:10.1001/jamadermatol.2017.1557
Dickey AK, Naik H, Keel SB, et al. Evidence-based consensus guidelines for the diagnosis and management of erythropoietic protoporphyria and X-linked protoporphyria. J Am Acad Dermatol 2023;89(6):1227-1237. doi:10.1016/j.jaad.2022.08.036
Drugs Mentioned In This Article





