

Minimal change disease causes abrupt onset of edema and heavy proteinuria, mostly in children. Kidney function is typically normal. Diagnosis is based on clinical findings or renal biopsy. Treatment is with corticosteroids or, in patients who do not respond, cyclophosphamide or cyclosporine. Prognosis is excellent. Minimal change disease causes abrupt onset of edema and heavy proteinuria, mostly in children. Kidney function is typically normal. Diagnosis is based on clinical findings or renal biopsy. Treatment is with corticosteroids or, in patients who do not respond, cyclophosphamide or cyclosporine. Prognosis is excellent.
(See also Overview of Nephrotic Syndrome.)
Minimal change disease is the most common cause of nephrotic syndrome in children > 1 year of age (70 to 90% of childhood nephrotic syndrome), but it also occurs in adults (15% of adult nephrotic syndrome) (1). The underlying disorder is almost always unknown, although rare cases may occur secondary to medication use (especially nonsteroidal anti-inflammatory drugs [NSAIDs]) and hematologic cancers (especially Hodgkin lymphoma).

General reference
1. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal Change Disease. Clin J Am Soc Nephrol 2017;12(2):332-345. doi:10.2215/CJN.05000516
Symptoms and Signs of Minimal Change Disease
Minimal change disease causes nephrotic syndrome, usually without hypertension or azotemia; microscopic hematuria occurs in more than half of adult patients with minimal change disease (1). Acute kidney injury can occur, particularly in patients > 50 to 60 years (2). Albumin is lost in the urine of patients with minimal change disease more so than larger serum proteins, probably because the disease causes changes in the charge barrier that affect albumin selectively.
Symptoms and signs references
1. Lionaki S, Mantios E, Tsoumbou I, et al. Clinical Characteristics and Outcomes of Adults with Nephrotic Syndrome Due to Minimal Change Disease. J Clin Med 2021;10(16):3632. doi:10.3390/jcm10163632
2. Meyrier A, Niaudet P. Acute kidney injury complicating nephrotic syndrome of minimal change disease. Kidney Int 2018;94(5):861-869. doi:10.1016/j.kint.2018.04.024
Diagnosis of Minimal Change Disease
In adults with idiopathic nephrotic syndrome, renal biopsy
In children, the diagnosis can be suspected (and treatment begun) based on the following typical characteristics:
Sudden onset of unexplained nephrotic-range proteinuria (≥ 3 g/day) that is mainly albumin
Normal kidney function
Non-nephritic urine sediment

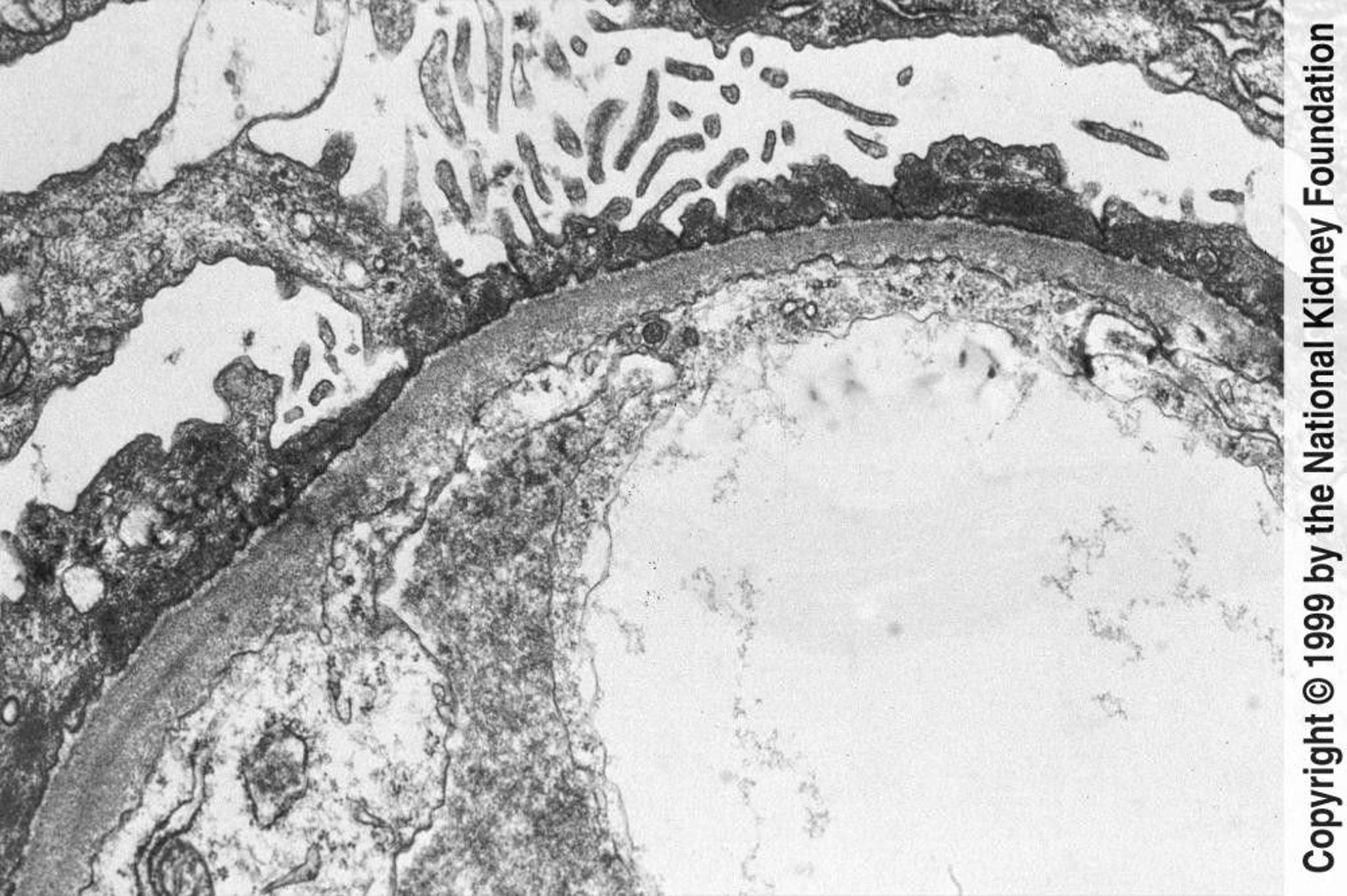
Diffuse effacement of foot processes can be seen on transmission electron microscopy (×800).
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
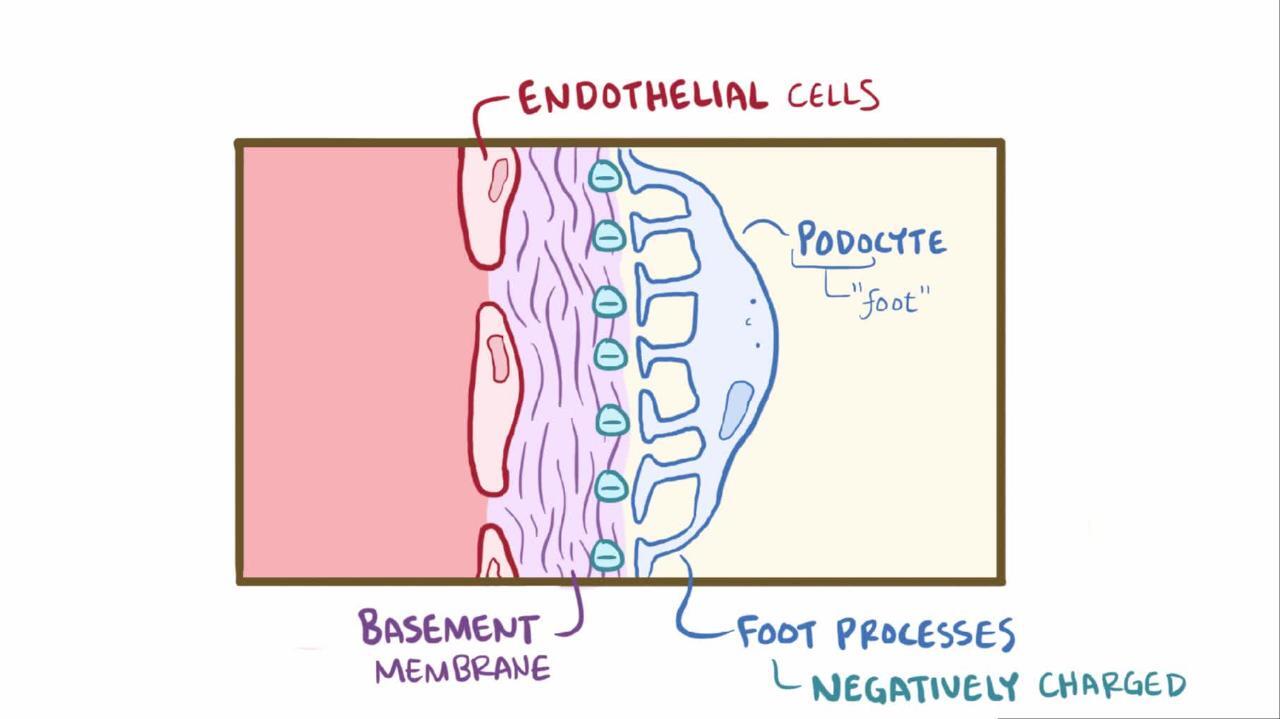
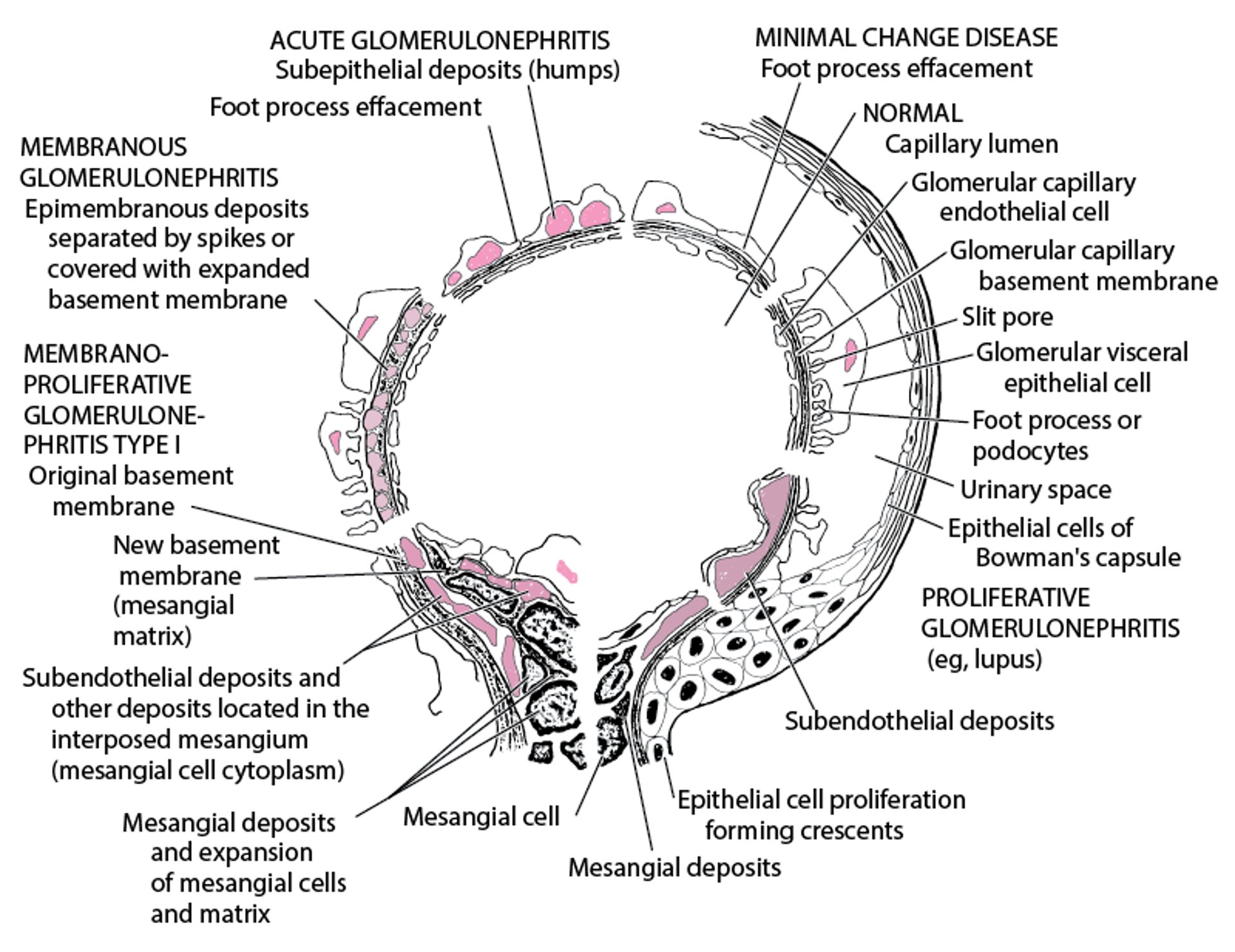
Renal biopsy is indicated in adults and in children with atypical presentations. Electron microscopy demonstrates edema with diffuse swelling (effacement) of foot processes of the epithelial podocytes (see figure ). Complement and Ig deposits are absent on immunofluorescence. Although effacement is not observed in the absence of proteinuria, heavy proteinuria may occur with normal foot processes.
Electron Microscopic Features in Immunologic Glomerular Disorders

Treatment of Minimal Change Disease
Corticosteroids
Sometimes cyclophosphamide, calcineurin inhibitors (eg, cyclosporine, tacrolimus), mycophenolate mofetil, or rituximabSometimes cyclophosphamide, calcineurin inhibitors (eg, cyclosporine, tacrolimus), mycophenolate mofetil, or rituximab
Loop diuretics (furosemide) and a low-sodium diet to manage swelling.Loop diuretics (furosemide) and a low-sodium diet to manage swelling.
Corticosteroids
Spontaneous remissions an occur in about one-third or more of cases, but most patients are given corticosteroids (1). Approximately 80 to 90% of patients respond to initial high-dose corticosteroid therapy (eg, prednisone 1 mg/kg daily for up to 8 weeks in adults), but the majority of responders relapse (). Approximately 80 to 90% of patients respond to initial high-dose corticosteroid therapy (eg, prednisone 1 mg/kg daily for up to 8 weeks in adults), but the majority of responders relapse (2). Patients who respond (ie, have cessation of proteinuria or a diuresis if edema is present) continue a more prolonged corticosteroid maintenance regimen. Treatment is usually continued for 1 to 2 years. More prolonged initial therapy and slower tapering of prednisone is associated with lower relapse rates (). Patients who respond (ie, have cessation of proteinuria or a diuresis if edema is present) continue a more prolonged corticosteroid maintenance regimen. Treatment is usually continued for 1 to 2 years. More prolonged initial therapy and slower tapering of prednisone is associated with lower relapse rates (3). Nonresponsiveness may be due to underlying focal sclerosis that was missed on biopsy due to sampling error.
Other immunosuppressive therapies
In corticosteroid nonresponders (< 5% of children and > 10% of adults), frequent relapsers, and corticosteroid-dependent patients, prolonged remission may be achieved with the addition of another immunosuppressive agent (4, 5). These agents include cyclophosphamide, a calcineurin inhibitor (cyclosporine or tacrolimus), mycophenolate mofetil, and possibly rituximab. The choice of agent is influenced by clinician and patient preferences, medication availability and cost, tolerability, and potential toxicity. ). These agents include cyclophosphamide, a calcineurin inhibitor (cyclosporine or tacrolimus), mycophenolate mofetil, and possibly rituximab. The choice of agent is influenced by clinician and patient preferences, medication availability and cost, tolerability, and potential toxicity.
Management of edema
Treatment references
1. Mak SK, Short CD, Mallick NP: Long-term outcome of adult-onset minimal-change nephropathy. Nephrol Dial Transplant 11(11):2192-2201, 1996. doi:10.1093/oxfordjournals.ndt.a027136
2. Larkins N, Kim S, Craig J, Hodson E: Steroid-sensitive nephrotic syndrome: an evidence-based update of immunosuppressive treatment in children. Arch Dis Child 101(4):404-408, 2016. doi:10.1136/archdischild-2015-308924
3. Mishra OP, Thakur N, Mishra RN, Prasad R: Prolonged versus standard prednisolone therapy for initial episode of idiopathic nephrotic syndrome. J Nephrol 25(3):394-400, 2012. doi:10.5301/jn.5000016
4. Larkins NG, Liu ID, Willis NS, et al: Non-corticosteroid immunosuppressive medications for steroid-sensitive nephrotic syndrome in children. Cochrane Database Syst Rev 4(4):CD002290, 2020. doi: 10.1002/14651858.CD002290.pub
5. Azukaitis K, Palmer SC, Strippoli GF, et al: Interventions for minimal change disease in adults with nephrotic syndrome. Cochrane Database Syst Rev 3(3):CD001537, 2022. doi: 10.1002/14651858.CD001537.pub5
6. Hampson KJ, Gay ML, Band ME: Pediatric Nephrotic Syndrome: Pharmacologic and Nutrition Management. Nutr Clin Pract 36(2):331–343, 2021. doi:10.1002/ncp.10622
7. Kalantar-Zadeh K, Fouque D: Nutritional Management of Chronic Kidney Disease. N Engl J Med 377(18):1765–1776, 2017. doi:10.1056/NEJMra1700312
8. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group: KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 100(4S):S1–S276, 2021. doi:10.1016/j.kint.2021.05.021
Prognosis for Minimal Change Disease
The prognosis for patients with minimal change disease who receive treatment is favorable, and > 80% of patients achieve remission. Progression to kidney failure occurs in < 5 % of patients and is more common among those who do not initially respond to corticosteroids (1).
Prognosis reference
1. Tarshish P, Tobin JN, Bernstein J, et al: Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J Am Soc Nephrol 8(5):769-776, 1997 doi: 10.1681/ASN.V85769
Key Points
Minimal change disease accounts for most cases of nephrotic syndrome in children and is usually idiopathic.
Suspect minimal change disease in children who have sudden onset of nephrotic-range proteinuria with normal kidney function and a non-nephritic urine sediment.
Confirm the diagnosis by renal biopsy in adults and atypical childhood cases.
Treat initially with corticosteroids.
The prognosis is favorable, and the majority of patients achieve remission with initial treatment.
Drug Information for the Topic





