



Primary malignant bone tumors are much less common than metastatic bone tumors, particularly in adults. Primary malignant bone tumors include osteosarcoma, adamantinoma, chondrosarcoma, chordoma, Ewing sarcoma of bone, and malignant giant cell tumor. (See also Overview of Bone and Joint Tumors and Overview of Leukemia.) Multiple myeloma and lymphoma of bone are also technically primary malignant bone tumors, but they are better discussed in the context of metastatic bone disease because the treatment principles are much more similar.
The 2 most widely used systems for staging these tumors are
The American Joint Committee on Cancer (AJCC) Cancer Staging Manual, Version 9: For osteosarcoma, chondrosarcoma, and Ewing sarcoma, staging is based on distinct tumor histology, histologic grade, maximal tumor size, nodal involvement, and metastases (TNM classification). The manual classifies tumors into 4 stages and is used for reporting cancer data. Notably, the updated staging system has a separate classification for pelvic and spinal sarcomas incorporating information regarding the number of pelvic and spinal segments involved.
The Musculoskeletal Tumor Society (MSTS) staging system: Used by orthopedic oncology surgeons based on histologic grade (eg, Stage I: low-grade histology and Stage II: high-grade histology, whether the tumor is contained entirely within the bone [A] or has broken outside of the cortex into surrounding soft tissue [B], and metastases Stage III). The typical (conventional) osteosarcoma with an associated soft tissue mass without metastases is Stage IIB in the MSTS system.
Osteosarcoma (osteogenic sarcoma)
Osteosarcoma is the most common malignant primary bone tumor (if one considers myeloma a marrow cell tumor and not a primary bone tumor) and is highly malignant. It is most common in children and teenagers, although it can occur at any age. There are 2 peaks of incidence; incidence is highest in adolescents and very young adults (coinciding with adolescent growth spurt) and secondary peak occurs in older adults (≥ age 60), especially in those with risk factors such as Paget disease, bone infarcts, and areas of bone previously exposed to high-dose radiation therapy for another cancer many years earlier. Children who carry the gene for hereditary retinoblastoma (variants of RB1 gene) and Li-Fraumeni syndrome (TP53 gene) are at higher risk of developing osteosarcoma.
Osteosarcoma produces malignant osteoid (immature bone) from tumor bone cells. Osteosarcoma usually develops around the knee (distal femur more often than proximal tibia) or in other long bones, particularly the metaphyseal-diaphyseal area, and may metastasize, usually to lung or other bone. Pain, swelling/mass, and loss of joint motion are the usual symptoms.


This radiograph of the knee shows a destructive osteosarcoma in the femur above the knee with a lytic destructive appearance and a classic Codman's triangle showing pathologic periosteal elevation.
Image courtesy of Lukas Nystrom, MD.
Findings on imaging studies vary and may include sclerotic or lytic features. Diagnosis of osteosarcoma requires biopsy. Patients need a chest CT to evaluate for lung metastases and a nuclear medicine whole-body bone scan to evaluate for bone metastases. MRI is done of the entire involved extremity to evaluate for metachronous lesions (or "skip lesions"). PET-CT may show distant metastases or metachronous lesions as well and is an evolving staging modality.
Treatment of osteosarcoma is a combination of chemotherapy and surgery. Use of adjuvant chemotherapy increases survival from < 20% to > 65% at 5 years for patients presenting with localized disease (1). Neoadjuvant chemotherapy begins before surgical resection. The goal is to treat the early micrometastatic disease assumed to be present even if it is not seen on staging imaging studies. Decreased peripheral soft tissue tumor mass (on imaging), increased mineralization (on radiograph), improved pain, and decreased serum alkaline phosphatase levels are all indicators of a favorable response. Ultimately, the response to chemotherapy is determined at the time of surgical resection with histologic mapping of the resected specimen by the pathologist. A positive prognostic finding is > 95% tumor necrosis on histologic mapping of the resected specimen by the pathologist. The goal of surgery is to resect the tumor—including all surrounding reactive tissue and a rim of surrounding normal tissue—en bloc, to avoid microscopic spillage of tumor cells. Necrosis and surgical margins are critically important to the patient's outcome. If a negative margin cannot be achieved with limb-salvage surgery, amputation should be considered.
More than 85% of patients can be treated with limb-sparing surgery without decreasing the long-term survival rate.
Continuation of chemotherapy after surgery is necessary. If there is nearly complete tumor necrosis (>90%) from preoperative chemotherapy, 5-year survival rate is reported to be 70 to 74% (2). Limited metastatic (eg, oligometastatic) disease to the lungs may be treated with thoracotomy and wedge resection of the lung lesion(s).
Variants of osteosarcoma that are different from conventional osteosarcoma and occur much less frequently include surface cortical lesions, such as parosteal osteosarcoma and periosteal osteosarcoma. Parosteal osteosarcomas most often involve the posterior cortex of the distal femur and usually are low-grade. Parosteal osteosarcomas require surgical en bloc resection but no chemotherapy if histology of the resected specimen confirms the tumor to be low-grade.
Periosteal osteosarcoma is more of a cartilage matrix surface tumor that also contains bone matrix and is malignant. It is often located on the mid-shaft femur and appears as a sunburst on radiograph. Likelihood of metastases for periosteal osteosarcomas is much greater than for parosteal osteosarcomas, but somewhat less than for typical osteosarcomas. Most of the time, periosteal osteosarcomas are treated similarly to conventional osteosarcomas with chemotherapy and surgical en bloc resection.
Adamantinoma
Adamantinoma is rare (< 1% of malignant bone tumors) and most often develops in the tibia, occasionally with involvement of the fibula. It can occur at any age, but it occurs most often in young and middle-aged individuals. Adamantinoma is a slow-growing malignancy that often manifests with pain and a palpable mass.

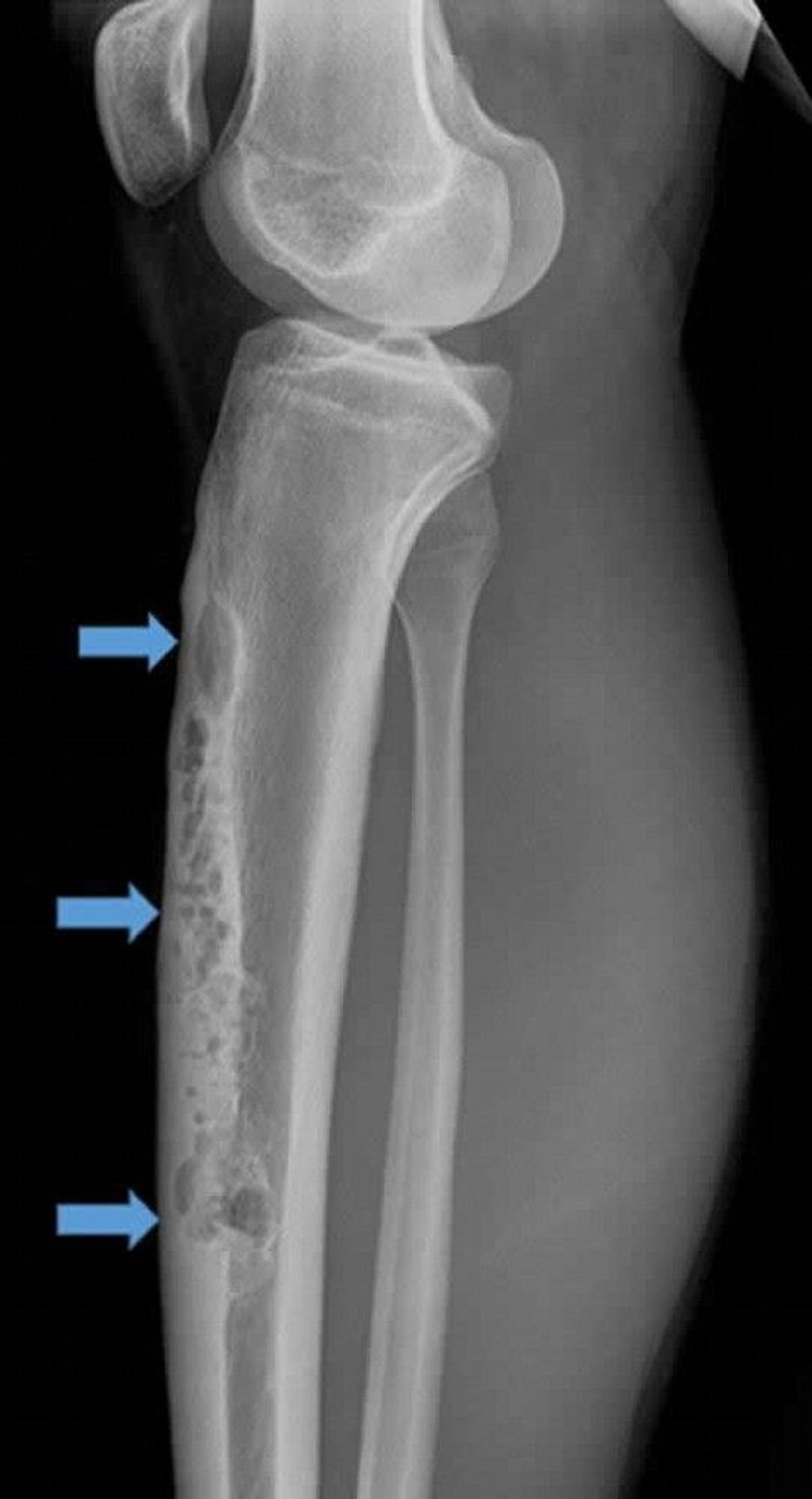
This lateral radiograph of right tibia shows a large anterior intracortical adamantinoma (arrows). Note the permeative, osteolytic "soap bubble" appearance.
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
The lesion typically manifests in the anterior crest of the tibia, and radiographs show a "soap bubble" osteolytic appearance. The histologic appearance is a biphasic pattern of nests of epithelial cells among a background of fibrous tissue. The tumor cells stain positive for keratin. The lesion can be confused with osteofibrous dysplasia of the anterior tibial cortex, which is a benign entity. It is likely that osteofibrous dysplasia of the anterior tibial cortex is on the same disease spectrum as adamantinoma (3), and may be a precursor lesion, but without the epithelial component characterizing it as malignant.
Metastases do occur, primarily to the lungs, but they are rare.
Treatment of adamantinoma consists of wide resection and reconstruction of the defect. Occasionally, amputation is necessary if negative surgical margins are not attainable or if the resulting defect is not reconstructible.
Chondrosarcoma
Chondrosarcomas are malignant cartilage-forming tumors. The majority of chondrosarcomas are primary tumors. They can also arise secondary and adjacent to an existing benign enchondroma or osteochondroma, particularly enchondromatosis (eg, in Ollier disease and Maffucci syndrome) or multiple hereditary exostoses (MHE). Chondrosarcomas tend to occur in middle-aged and older adults. They may develop in flat bones (eg, pelvis, scapula) or any long bone (most often femur and humerus among the long bones) and can have a component extending into surrounding soft tissues.

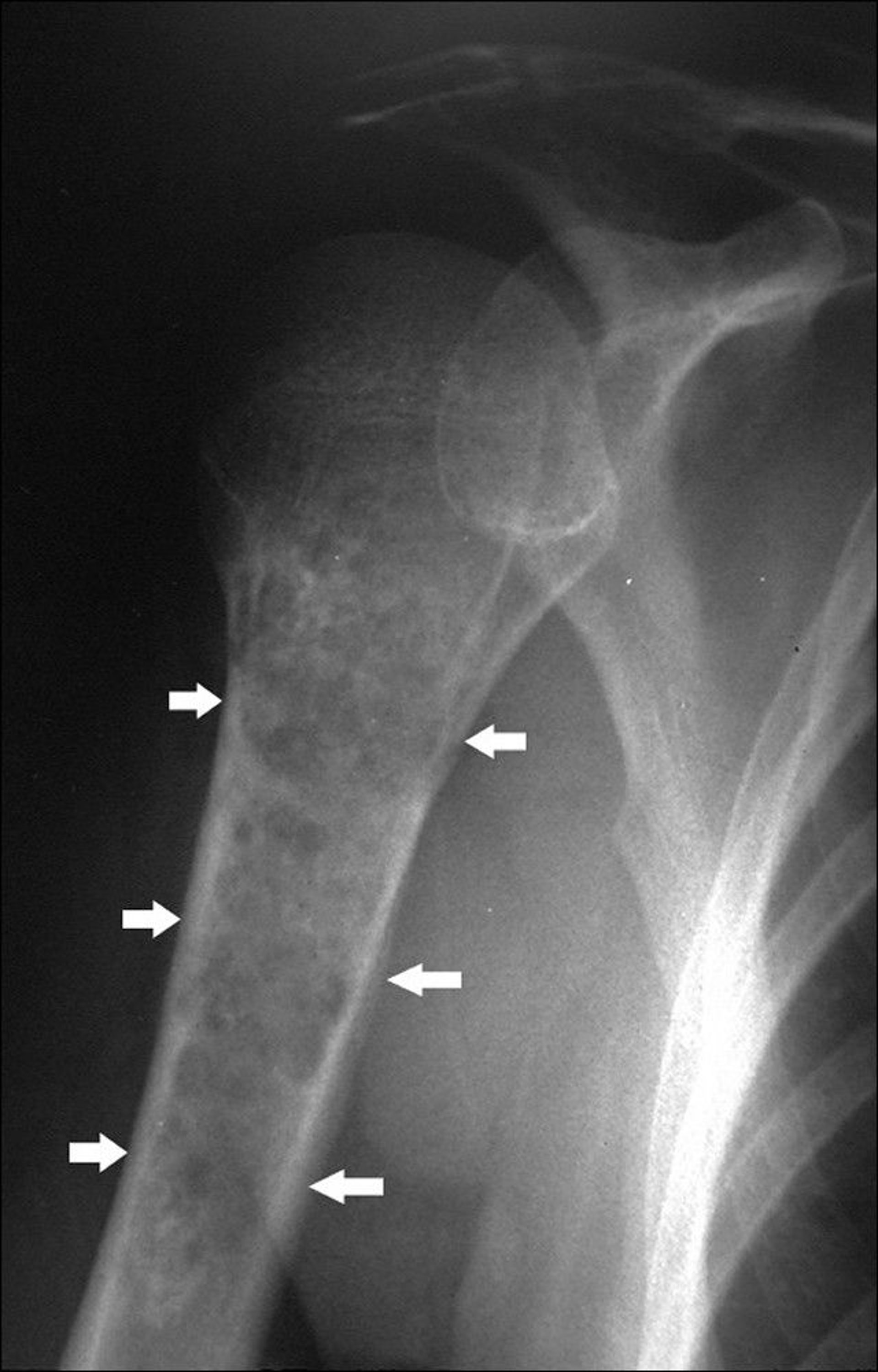
This shoulder radiograph shows a stippled calcified lesion in the humerus (arrows) with endosteal scalloping, including some areas of destruction through the cortex.
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Radiographs often reveal punctate calcifications within the matrix of the tumor. The amount of calcification can vary. Chondrosarcomas often also exhibit cortical bone destruction and loss of normal bone trabeculae. MRI can nicely demonstrate the complete extent of marrow involvement and also demonstrates the soft tissue mass, if present. Bone scan should be done to complete the staging evaluation, looking for other potential sites of osseous disease. While chondrosarcoma is often readily diagnosed based on radiographic findings alone, biopsy is beneficial when the diagnosis is in question and can also aid in determination of the tumor grade (probability of metastasizing) if that information is helpful to inform treatment alternatives. However, it should be noted that it is often difficult to differentiate low-grade chondrosarcomas from enchondromas by imaging and histology (4).
Low-grade intramedullary chondrosarcomas (grade 1) are often treated with intralesional curettage with the addition of an adjuvant (thermal or chemical). Some surgeons prefer surgical en bloc resection for low-grade tumors to reduce risk of recurrence. Intermediate or high-grade tumors require more aggressive treatment in the form of a wide resection. When surgical resection with maintenance of function is not possible, or if an adequate margin cannot be achieved, amputation may be necessary. There are no other therapies thought to be effective against chondrosarcoma. Because of the potential to implant the tumor, meticulous care must be taken to avoid spillage of tumor cells into the soft tissues during a biopsy or surgery. Recurrence is inevitable if tumor cells spill. If no spillage occurs, the cure rate depends on the tumor grade. Low-grade tumors are nearly all cured with adequate treatment. Because these tumors have limited vascularity, chemotherapy and radiation therapy have little efficacy.
Chordoma
Chordoma is a rare malignancy that develops from the remnants of the primitive notochord. It tends to occur at the ends of the spinal column, usually in the middle of the sacrum or near the base of the skull. A chordoma in the sacrococcygeal region typically causes substantial pain. A chordoma in the base of the skull can cause deficits in a cranial nerve, most commonly in the optic nerve. Symptoms of chordoma may exist for months to several years before diagnosis.

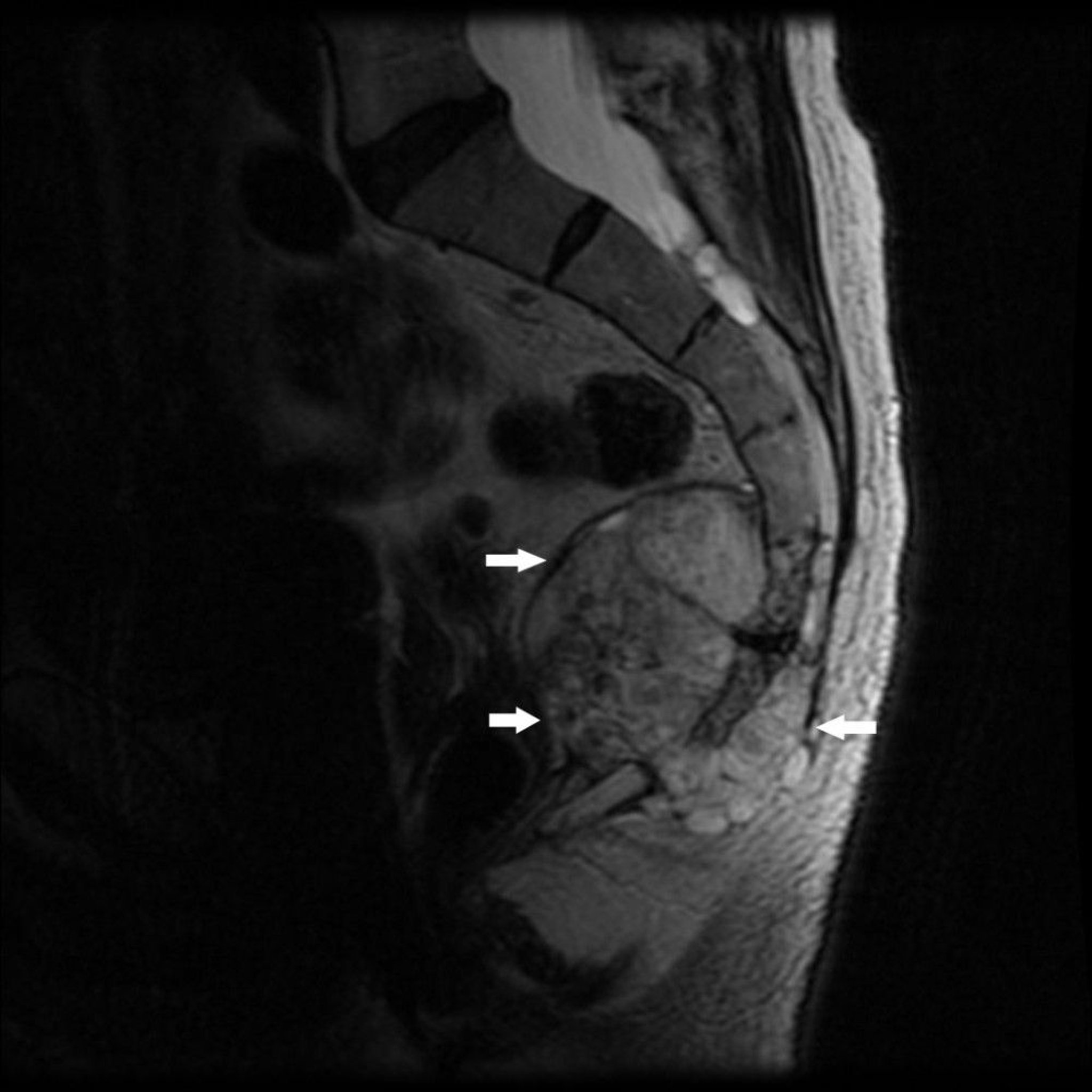
This MRI shows a tumor at the caudal end of the sacrum (S4) and coccyx with bone destruction and soft tissue mass (arrows), typical of a chordoma.
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
A chordoma appears on imaging studies as a destructive bone lesion that may be associated with a soft tissue mass. It is best visualized by MRI or CT given the predominant soft tissue component. Biopsy is required to confirm the diagnosis. The pathognomonic cell type is the physaliferous cell; these cells are characterized by being very large and having a small round nucleus and bubbly appearing cytoplasm.
Chordomas in the sacrococcygeal region may be cured by radical en bloc excision, with or without adjuvant radiotherapy. The surgery almost certainly requires sacrifice of all nerve roots at the level of involvement and below. Depending on the level of the tumor involvement, the proposed surgery carries substantial morbidity of loss of bowel function, bladder function, and/or sexual function (5). If surgery is felt to be excessively morbid, curative-intent radiotherapy can be offered in its place. Chordomas in the base of the skull are usually inaccessible to surgery but may respond to radiation therapy. The rate of local recurrence is high after tumor resection. Metastasis, although less common, may occur.
Ewing sarcoma of bone
Ewing sarcoma of bone is a small blue round-cell bone tumor with a peak incidence between 10 and 20 years. Ewing sarcoma is related to peripheral primitive neuroectodermal tumors (PNET) and Askin malignant small cell tumor of the chest wall, which are now considered part of the Ewing sarcoma family of tumors. Most tumors develop in the extremities, but any bone may be involved. Ewing sarcoma most often involves the diaphyseal region with some extension into the metaphyseal region. Pain and swelling are the most common symptoms.

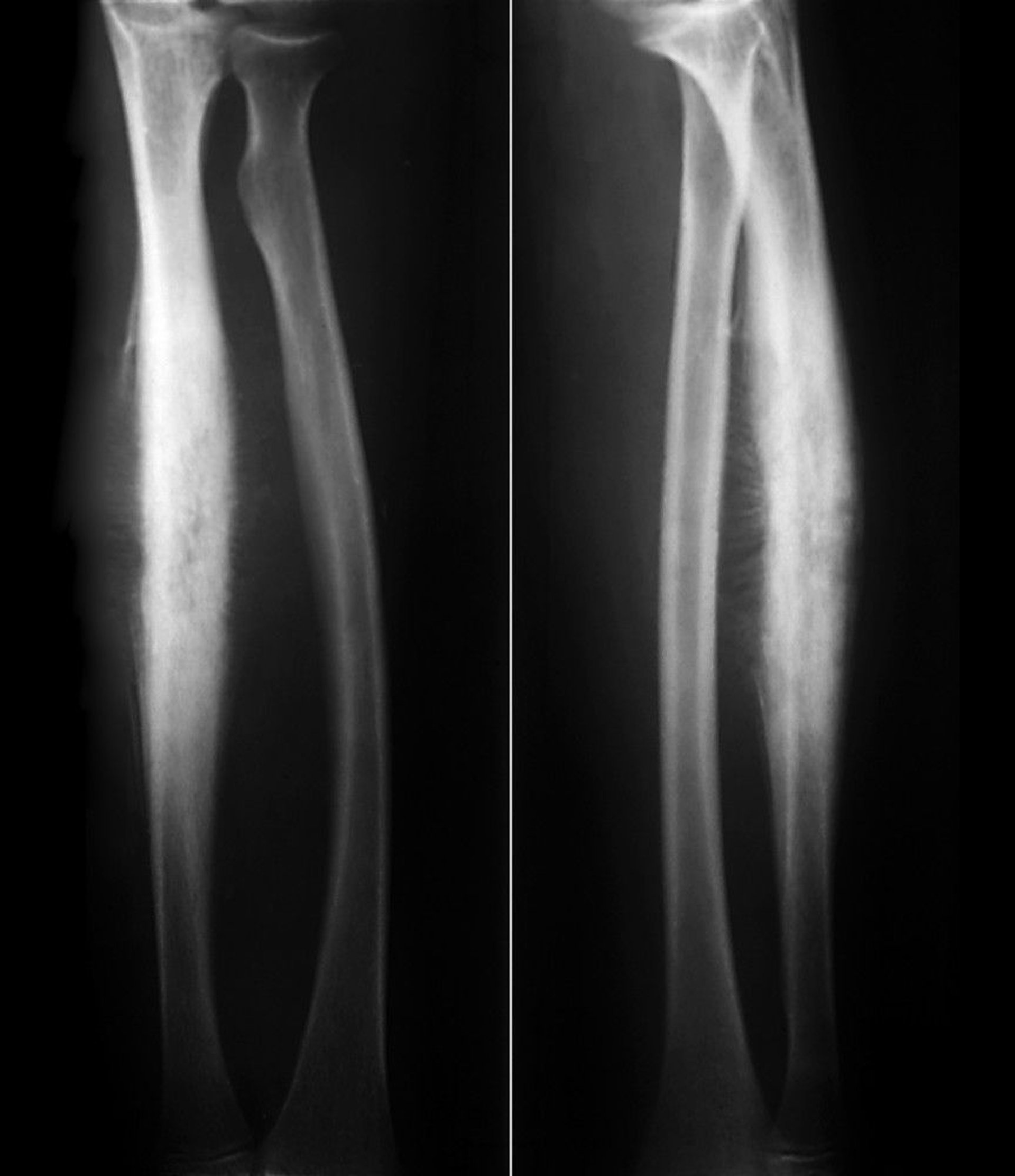
This Ewing sarcoma of the radial shaft shows subperiosteal reactive new bone formation in a classic "sunburst" periosteal reaction along with lytic destruction of cortical bone.
ZEPHYR/SCIENCE PHOTO LIBRARY
Lytic destruction, particularly a permeative pattern, is the most common finding on imaging, but multiple layers of subperiosteal reactive new bone formation may give an onion-skin appearance. Radiographs classically do not usually reveal the full extent of bone involvement, and a large soft tissue mass usually surrounds the affected bone. MRI better defines disease extent, which can help guide treatment.
Many other benign and malignant tumors can appear very similarly, so diagnosis of Ewing sarcoma is made by biopsy. At times this type of tumor may be confused with an infection. Accurate histologic diagnosis can be accomplished with cytogenetic analysis and identification of molecular markers, including evaluation for a typical chromosomal translocation between chromosomes 11 and 22 resulting in the EWSR1-FLI1 fusion product. This fusion product is present in approximately 85% of cases; however, several different structural translocations in different fusion gene patterns have been identified (6).

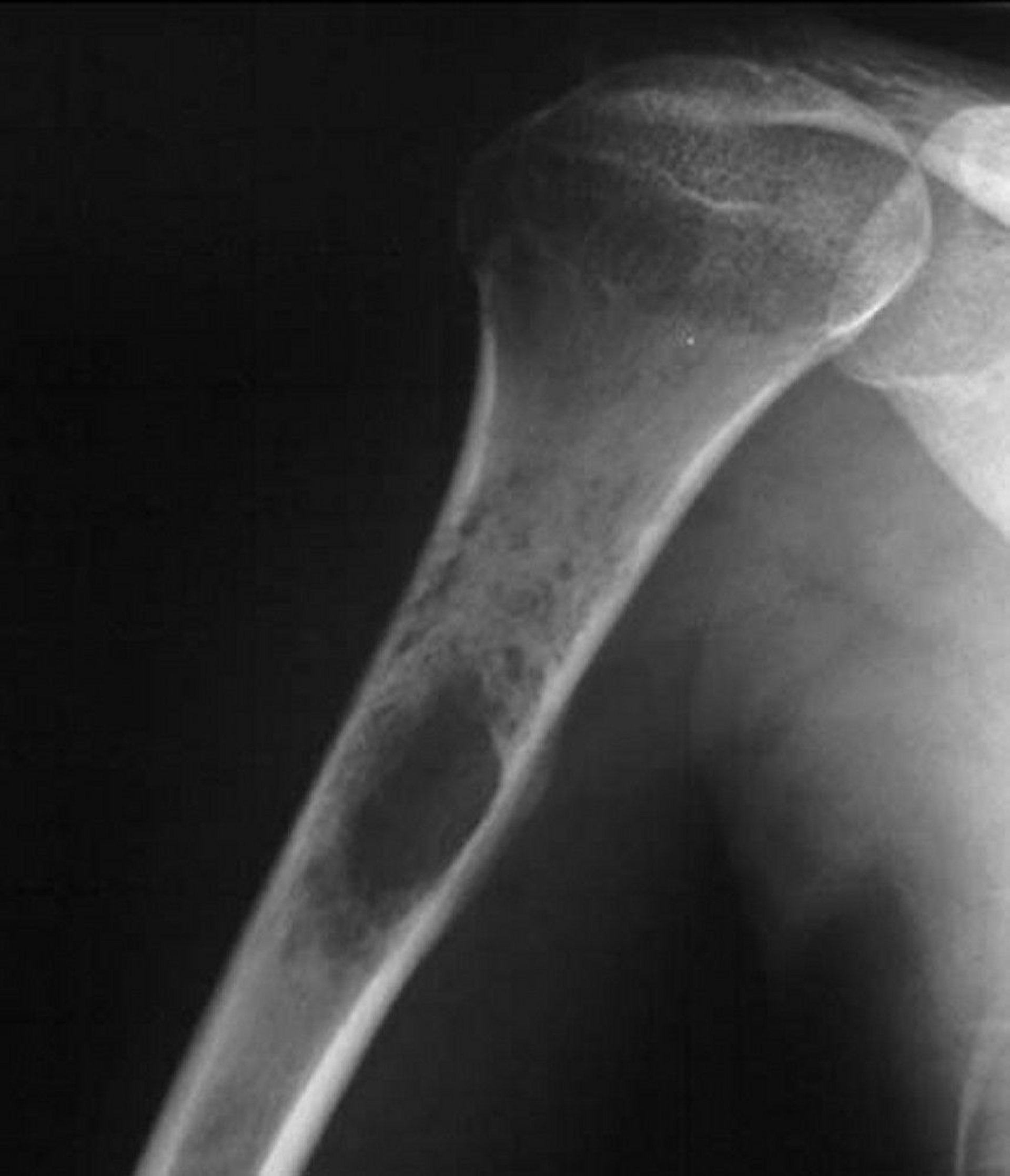
This shoulder radiograph shows a permeative destructive tumor arising in the proximal humerus, which is typical of Ewing sarcoma.
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Treatment of Ewing sarcoma includes chemotherapy and surgery with or without radiation therapy. Currently, > 60% of patients with primary localized Ewing sarcoma may be cured by this multimodal approach (7). Cure is sometimes possible even with metastatic disease. Chemotherapy in conjunction with surgical en bloc resection, if possible, has a lower rate of local relapse than chemotherapy in conjunction with radiation therapy, although these differences are small and must be weighed against the morbidity of surgical resection. Most often, surgery is favored when possible.
Lymphoma of bone
Lymphoma of bone primarily affects adults. The typical subtype is diffuse large B-cell lymphoma. It may arise in any bone. The tumor consists primarily of small round cells, often with a mixture of reticular cells, lymphoblasts, and lymphocytes. It may develop as an isolated primary bone tumor, in association with similar tumors in other tissues, or as a metastasis from known soft tissue lymphomatous disease. Pain and swelling are the usual symptoms of lymphoma of bone. Pathologic fracture may be a presenting symptom.

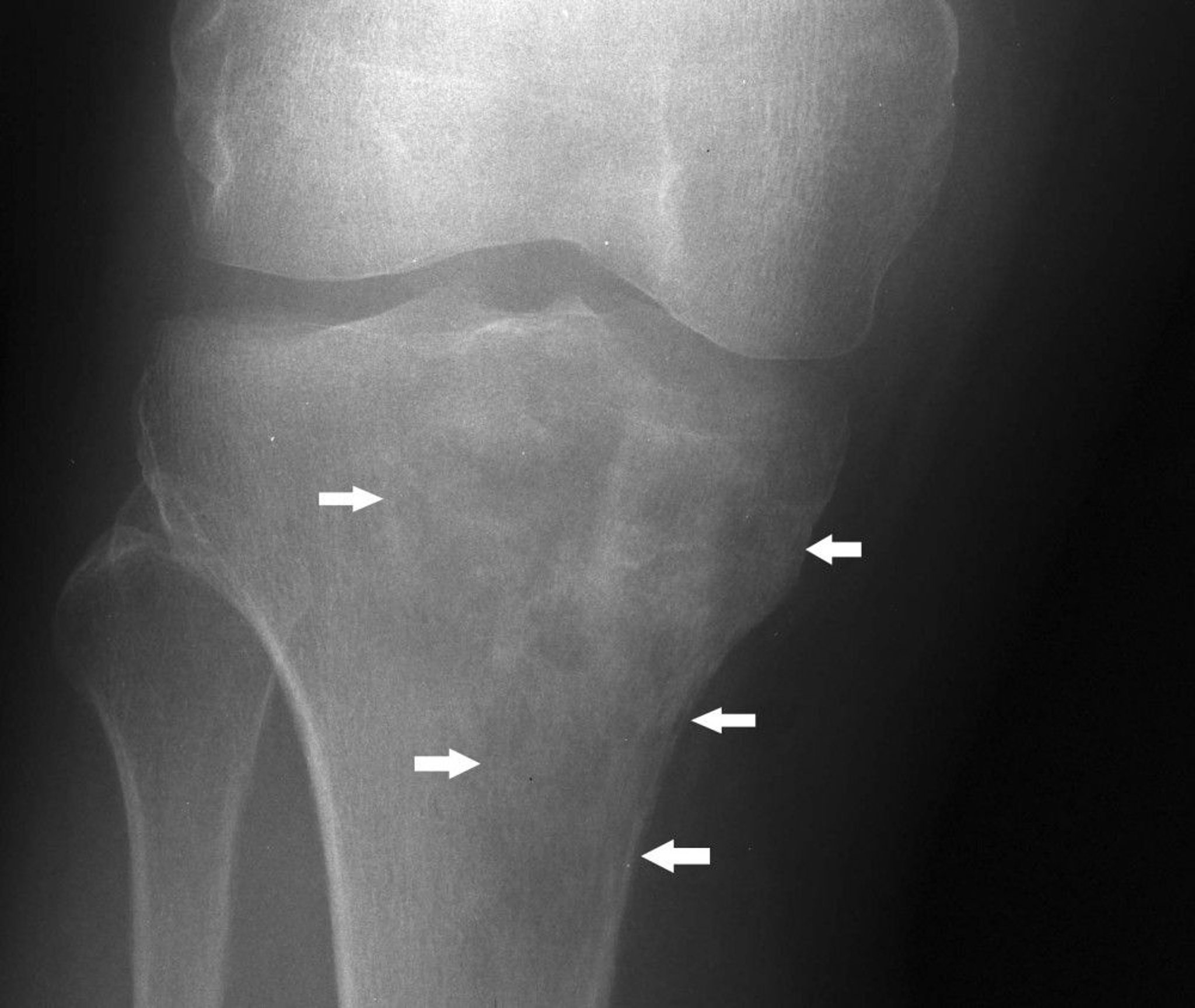
This knee radiograph shows lymphoma in the proximal tibia with a mixed lytic and sclerotic appearance below the medial tibial condyle (arrows).
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Imaging studies reveal bone destruction, which may be in a mottled or patchy or even infiltrating, permeative pattern, often with a clinical and radiographic large soft tissue mass. In many cases, the extent of soft tissue and marrow involvement noted on MRI will be very significant in comparison to changes seen on radiographs. However, in advanced disease, the entire outline of the affected bone may be lost.
The most common histologic subtype is diffuse large B-cell lymphoma. In isolated primary bone diffuse large B-cell lymphoma, the 5-year survival rate is 88% (8).
Bone lymphomas are typically treated with systemic chemotherapy. Radiation therapy can be used as an adjuvant in some cases if inadequate radiographic response is seen after chemotherapy. Stabilization of long bones may be necessary to prevent or treat a pathologic fracture. On occasion, arthroplasty is required to treat a nonsalvageable pathologic fracture near the end of the bone
References
1. Link MP, Goorin AM, Horowitz M, et al. Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin Orthop Relat Res. 1991;(270):8-14.
2. Richardson SM, Wurtz LD, Collier CD. Ninety Percent or Greater Tumor Necrosis Is Associated With Survival and Social Determinants of Health in Patients With Osteosarcoma in the National Cancer Database. Clin Orthop Relat Res. 2023 Mar 1;481(3):512-522. doi: 10.1097/CORR.0000000000002380. Epub 2022 Sep 13. PMID: 36099400; PMCID: PMC9928876.
3. Nascimento AF, Kilpatrick SE, Reith JD. Osteofibrous Dysplasia and Adamantinoma. Surg Pathol Clin. 2021;14(4):723-735. doi:10.1016/j.path.2021.06.012
4. Skeletal Lesions Interobserver Correlation among Expert Diagnosticians (SLICED) Study Group. Reliability of histopathologic and radiologic grading of cartilaginous neoplasms in long bones. J Bone Joint Surg Am. 2007;89(10):2113-2123. doi:10.2106/JBJS.F.01530
5. Moran D, Zadnik PL, Taylor T, et al. Maintenance of bowel, bladder, and motor functions after sacrectomy. Spine J. 2015;15(2):222-229. doi:10.1016/j.spinee.2014.08.445
6. Riggi N, Suvà ML, Stamenkovic I. Ewing's Sarcoma. Reply. N Engl J Med. 2021;384(15):1477-1478. doi:10.1056/NEJMc2102423
7. Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108-3114. doi:10.1200/JCO.2000.18.17.3108
8. Beal K, Allen L, Yahalom J. Primary bone lymphoma: treatment results and prognostic factors with long-term follow-up of 82 patients. Cancer. 2006 Jun 15;106(12):2652-6. doi: 10.1002/cncr.21930. PMID: 16700039.





