

Marfan syndrome is a genetic connective tissue disorder of the fibrillin-1 protein that leads to ocular, musculoskeletal, and cardiovascular abnormalities (eg, dilation of ascending aorta, which can lead to aortic dissection). Diagnosis is clinical and based on genetic testing. Treatment may include beta-blockers and/or angiotensin II receptor blockers to slow dilation of the ascending aorta, and prophylactic aortic surgery.). Diagnosis is clinical and based on genetic testing. Treatment may include beta-blockers and/or angiotensin II receptor blockers to slow dilation of the ascending aorta, and prophylactic aortic surgery.
Marfan syndrome is an autosomal dominant, highly penetrant, progressive genetic condition caused by pathogenic variants in the FBN1 gene encoding the glycoprotein fibrillin-1, which is the main component of microfibrils and helps anchor cells to the extracellular matrix. The principal structural defect involves the cardiovascular, musculoskeletal, and ocular systems. The pulmonary system and central nervous system may also be affected.
Neonatal Marfan syndrome is a severe form of the disease that manifests before 1 year of age and is caused by a specific subset of FBN1 mutation (1). This form is characterized by significant valvular disease and heart failure.
There are many different manifestations of the genetic mutation that causes Marfan syndrome; however, it is typically recognized by the constellation of long limbs, aortic root dilation, and dislocated lenses.
The estimated prevalence of Marfan syndrome is approximately 1.5 to 6.8/100,000 people (2, 3).
General references
1. van der Leest EC, van der Hulst AE, Pals G, et al. Genotype-Phenotype Correlations, Treatment, and Prognosis of Children With Early-Onset (Neonatal) Marfan Syndrome. Clin Genet. 2025;108(2):134-145. doi:10.1111/cge.14722
2. Milewicz DM, Braverman AC, De Backer J, et al. Marfan syndrome [published correction appears in Nat Rev Dis Primers. 2022 Jan 17;8(1):3. doi: 10.1038/s41572-022-00338-w.]. Nat Rev Dis Primers. 2021;7(1):64. Published 2021 Sep 2. doi:10.1038/s41572-021-00298-7
3. Groth KA, Hove H, Kyhl K, et al. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J Rare Dis. 2015;10:153. Published 2015 Dec 2. doi:10.1186/s13023-015-0369-8
Symptoms and Signs of Marfan Syndrome
Cardiovascular system
Major findings include:
Aortic root dilation, ascending aortic aneurysm, or aortic dissection
Valvular prolapse (typically mitral)
Cardiovascular complications result from pathologic changes in the aortic root and ascending aorta, preferentially affecting the medial layer in areas subject to the greatest hemodynamic stress. The aorta progressively dilates, usually beginning in the sinuses of the aortic valve, sometimes before 10 years of age. The majority of patients with Marfan syndrome, including children, have aortic root dilation, which can cause aortic regurgitationand the characteristic diastolic decrescendo murmur over the aortic area or apex (1). Progressive aortic root dilation increases the risk of aortic dissection, the primary life-threatening complication of Marfan syndrome.



This 3D, colored CT angiogram shows an ascending aortic aneurysm with dissection.
VSEVOLOD ZVIRYK/SCIENCE PHOTO LIBRARY
Redundant cusps and chordae tendineae may lead to mitral valve prolapse or regurgitation; mitral valve prolapse may cause a midsystolic click with a late systolic or holosystolic murmur, signifying regurgitation.
Bacterial endocarditis may develop in the affected valves.
Musculoskeletal system
Patients are taller than average for age and family; arm span exceeds height. Arachnodactyly (disproportionately long, thin digits) is noticeable, often by the thumb sign (the distal phalanx of the thumb protrudes beyond the edge of the clenched fist). The chest wall abnormalities pectus carinatum (outward displacement) and pectus excavatum (inward displacement) are common as are joint hyperflexibility, limited elbow extension (though usually small flexion contractures to the elbows), genu recurvatum (backward curvature of the legs at the knees), pes planus (flat feet) or hindfoot valgus, kyphoscoliosis, and diaphragmatic and inguinal hernias. Subcutaneous fat usually is sparse.
Facial features may include dolichocephaly, enophthalmos, down-slanting palpebral fissures, retrognathia, and malar hypoplasia. The palate is often high-arched.
Skin may exhibit striae (stretch marks).
Severity of musculoskeletal findings varies greatly among patients.

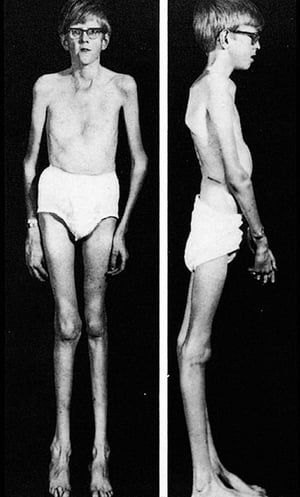
This image shows the typical body habitus in an adolescent with Marfan syndrome, including kyphoscoliosis, pectus excavatum, and genu recurvatum.
This image shows the typical body habitus in an adolescent with Marfan syndrome, including kyphoscoliosis, pectus excav
By permission of the publisher. From Macro R: Atlas of Heart Diseases: Congenital Heart Disease. Edited by E Braunwald (series editor) and RM Freedom. Philadelphia, Current Medicine, 1997.

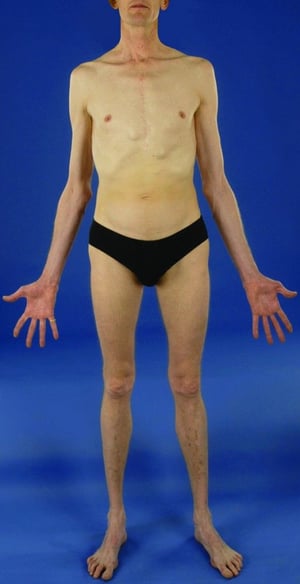
This patient with Marfan syndrome is taller than his family, and his arm span exceeds his height. Note also the incomplete extension of the elbows.
This patient with Marfan syndrome is taller than his family, and his arm span exceeds his height. Note also the incompl
© Springer Science+Business Media

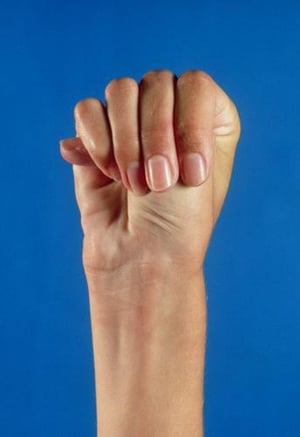
The thumb protrudes beyond the edge of the clenched fist in this photo of a person with Marfan syndrome.
The thumb protrudes beyond the edge of the clenched fist in this photo of a person with Marfan syndrome.
MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY


Marfan syndrome is characterized by abnormally long digits. In this photo, the woman's thumb overlaps her second digit when her hand is wrapped around her wrist.
Marfan syndrome is characterized by abnormally long digits. In this photo, the woman's thumb overlaps her second digit
Photo courtesy of David D. Sherry, MD.


SCIENCE PHOTO LIBRARY


The photo on the left shows a person with a pectus excavatum (funnel chest).The photo on the right shows a person with a pectus carinatum (pigeon chest).
The photo on the left shows a person with a pectus excavatum (funnel chest).The photo on the right shows a person with
DR P. MARAZZI / SCIENCE PHOTO LIBRARY
This image shows the typical body habitus in an adolescent with Marfan syndrome, including kyphoscoliosis, pectus excavatum, and genu recurvatum.
This image shows the typical body habitus in an adolescent with Marfan syndrome, including kyphoscoliosis, pectus excav
By permission of the publisher. From Macro R: Atlas of Heart Diseases: Congenital Heart Disease. Edited by E Braunwald (series editor) and RM Freedom. Philadelphia, Current Medicine, 1997.
This patient with Marfan syndrome is taller than his family, and his arm span exceeds his height. Note also the incomplete extension of the elbows.
This patient with Marfan syndrome is taller than his family, and his arm span exceeds his height. Note also the incompl
© Springer Science+Business Media
The thumb protrudes beyond the edge of the clenched fist in this photo of a person with Marfan syndrome.
The thumb protrudes beyond the edge of the clenched fist in this photo of a person with Marfan syndrome.
MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY
Marfan syndrome is characterized by abnormally long digits. In this photo, the woman's thumb overlaps her second digit when her hand is wrapped around her wrist.
Marfan syndrome is characterized by abnormally long digits. In this photo, the woman's thumb overlaps her second digit
Photo courtesy of David D. Sherry, MD.
SCIENCE PHOTO LIBRARY
The photo on the left shows a person with a pectus excavatum (funnel chest).The photo on the right shows a person with a pectus carinatum (pigeon chest).
The photo on the left shows a person with a pectus excavatum (funnel chest).The photo on the right shows a person with
DR P. MARAZZI / SCIENCE PHOTO LIBRARY
Ocular system
Findings include ectopia lentis (subluxation or upward dislocation of the lens) and iridodonesis (tremulousness of the iris). The margin of the dislocated lens can often be seen through the undilated pupil.
High-grade myopia may be present, and spontaneous retinal detachment may occur.
Pulmonary system
Cystic lung disease and recurrent spontaneous pneumothorax may occur. These disorders can cause chest pain and shortness of breath.
Central nervous system
Dural ectasia (widening of the dural sac surrounding the spinal cord) is a common finding and most frequently occurs in the lumbosacral spine. It may cause headache, lower back pain, or neurologic deficits manifested by bowel or bladder weakness.
Symptoms and signs reference
1. Wozniak-Mielczarek L, Sabiniewicz R, Drezek-Nojowicz M, et al. Differences in Cardiovascular Manifestation of Marfan Syndrome Between Children and Adults. Pediatr Cardiol. 2019;40(2):393-403. doi:10.1007/s00246-018-2025-2
Diagnosis of Marfan Syndrome
Clinical criteria
Genetic testing
Echocardiography/MRI (measurement of the aortic root, detection of valve prolapse)
Ophthalmologic examination (lens abnormalities)
Spinal and pelvic radiographs
MRI of the lumbosacral spine (dural ectasia)
The diagnosis can be difficult, particularly in the absence of a family history or a known pathogenic genetic mutation, because many patients have only a few typical symptoms and signs; furthermore, children and adolescents may have an evolving phenotype. Additionally, 25% of patients with Marfan syndrome have de novo mutations and therefore do not have a positive family history.
Diagnosis of Marfan syndrome requires the following criteria (1):
Aortic root dilation with FBN1 mutation, ectopia lentis, or systemic score > 7
FBN1 mutation and ectopia lentis
Family history of Marfan syndrome with ectopia lentis, aortic root dilation, or systemic score > 7
Genetic testing is highly sensitive. The FBN1 mutation is found in 95% of children with Marfan syndrome (2).
A detailed physical examination, focused on the elements of the systemic score, is performed. This examination includes an ophthalmologic evaluation with a focus on myopia and ectopia lentis.
Imaging tests are required to detect aortic root dilation and some elements of the systemic score (see table ).
Echocardiography or cardiac MRI is performed to evaluate for aortic root dilation, which is indexed to the patient's body size; the degree of dilation required for diagnosis depends on the patient's family history and age:
No family history: Aortic root Z-score ≥ 2
Positive family history: Aortic root Z-score ≥ 2 for patients > 20 years of age or Z-score ≥ 3 for those < 20 years of age
The systemic score is based on the physical and radiographic examinations (see table ) (1, 3, 4).


This radiograph shows scoliosis.
A Cobb angle of 20° is required to count towards the systemic score when evaluating for Marfan syndrome.
MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY
Systemic Score for Marfan Syndrome
Criteria | Points* |
|---|---|
Wrist or thumb sign | 1 for either or 3 for both |
Pectus carinatum | 2 |
Pectus excavatum or chest asymmetry | 1 |
Hindfoot valgus | 2 |
Pes planus | 1 |
Spontaneous pneumothorax | 2 |
Dural ectasia (requires lumbar spine MRI) | 2 |
Protrusio acetabuli (requires pelvic radiograph) | 2 |
Scoliosis or thoracolumbar kyphosis (requires spinal radiograph) | 1 |
Reduced elbow extension | 1 |
3 of 5 characteristic facial features (dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, and retrognathia) | 1 |
Skin striae | 1 |
Severe myopia (prescription > 3 diopters) | 1 |
Mitral valve prolapse (requires echocardiography) | 1 |
Reduced upper/lower segment ratio and increased arm span/height ratio† | 1 |
* A score of ≥ 7 points is considered positive. | |
† Not accurate in the presence of severe scoliosis or kyphosis. | |
Data from Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-485. doi:10.1136/jmg.2009.072785. | |
Genetic testing for FBN1 mutations can help establish the diagnosis in people who do not meet all clinical criteria, but FBN1 mutation-negative patients exist. Prenatal diagnosis by analysis of the FBN1 gene is hampered by poor genotype/phenotype correlation (> 1700 different mutations have been described).
The differential diagnosis of Marfan syndrome includes Loeys-Dietz syndrome, vascular type Ehlers-Danlos syndrome, hereditary thoracic aortic disease, and congenital contractural arachnodactyly. Homocystinuria can partially mimic Marfan syndrome but can be differentiated by detecting homocystine in the urine.
(See also Aortic Root Z-Scores for Children, Aortic Root Z-Scores for Adults, and Calculation of Systemic Score.)
Diagnosis references
1. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-485. doi:10.1136/jmg.2009.072785
2. Morris SA, Flyer JN, Yetman AT, et al. Cardiovascular Management of Aortopathy in Children: A Scientific Statement From the American Heart Association. Circulation. 2024;150(11):e228-e254. doi:10.1161/CIR.0000000000001265
3. The Marfan Foundation. Summary of Diagnostic Criteria. Accessed May 12, 2025.
4. The Marfan Foundation. Calculation of Systemic Score. Accessed May 12, 2025.
Treatment of Marfan Syndrome
Beta-blockers and/or angiotensin II receptor blockersBeta-blockers and/or angiotensin II receptor blockers
Elective aortic repair and valve repair
Bracing and surgery for scoliosis
Vision correction, lensectomy, and laser surgery
Sports and exercise guidance
Treatment of Marfan syndrome is focused on prevention and treatment of complications, particularly aortic root dissection.
Cardiovascular, musculoskeletal, and ocular findings should be reevaluated annually.
Appropriate genetic counseling is indicated for reproductive purposes.
Medications
All adult patients should routinely be given beta-blockers (eg, atenolol, propranolol), or angiotensin II receptor blockers (eg, losartan, irbesartan), or a combination of both at a maximally tolerated dose to help slow aortic dilation and prevent cardiovascular complications (All adult patients should routinely be given beta-blockers (eg, atenolol, propranolol), or angiotensin II receptor blockers (eg, losartan, irbesartan), or a combination of both at a maximally tolerated dose to help slow aortic dilation and prevent cardiovascular complications (1).
For children, monotherapy is started when aortic root diameter is abnormal (Z-score ≥ 2 for patients < 16 years old, and absolute diameter ≥ 3.5 cm for patients ≥ 16 years old); dual therapy is started as dilation progresses (2).
These medications lower myocardial contractility and blood pressure, potentially affect signalling pathways that are implicated in the pathogenesis of disease, and reduce progression of aortic root dilation and risk of dissection (3).
Surgeries
Prophylactic surgery (aortic root replacement) is offered to adults when the aortic diameter is > 5.0 cm (> 4.5 cm in patients with certain high-risk features) (1). For children, the threshold diameter is based on age as well as risk profile (2). Pregnant patients are at especially high risk of aortic complications; elective aortic repair before conception should be discussed. Severe valve regurgitation is also surgically repaired.
Personalized external aortic root support (PEARS) is a surgical option that reinforces but does not require replacement of the aortic root (4).
Scoliosis is managed with bracing as long as possible, but surgical intervention is encouraged in patients with spinal curvature of 40 to 50°.
Ocular manifestations, such as lens dislocation, retinal detachment, and glaucoma, require surgical correction. For a dislocated lens, correction may involve removal of the lens (lensectomy) followed by a replacement intraocular lens, or vision correction if the lens is not replaced. These interventions are highly successful because of advancements in ocular technologies.
Physical activity
Sports and exercise guidance for patients with Marfan syndrome is focused on risk assessment and shared decision making (2, 5).
For older adolescents and adults with Marfan syndrome, particularly those with aortic root dilation, exercising at peak intensity, training requiring a forceful Valsalva maneuver (such as heavy weightlifting), and sports with a risk of collision or bodily contact are discouraged. These risks are balanced with the benefits of sports participation and physical activity.
Joint hypermobility and ocular complications are additional considerations for individualized sports and exercise counseling.
Pregnancy
The risks of pregnancy, labor, and delivery should be discussed with all patients with Marfan syndrome and aortopathy who might become pregnant, ideally before conception. For some, prepregnancy elective aortic repair may be offered.
Specific recommendations for delivery are based on aortic diameter (1):
Aortic diameter < 4.0 cm: Vaginal delivery (when otherwise appropriate)
Aortic diameter 4.0 to 4.5 cm: Consider regional anesthesia for vaginal delivery, expedited second stage, assisted delivery, or cesarean delivery
Aortic diameter > 4.5 cm: Cesarean delivery
Treatment references
1. Writing Committee Members, Isselbacher EM, Preventza O, et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;80(24):e223-e393. doi:10.1016/j.jacc.2022.08.004
2. Morris SA, Flyer JN, Yetman AT, et al. Cardiovascular Management of Aortopathy in Children: A Scientific Statement From the American Heart Association. Circulation. 2024;150(11):e228-e254. doi:10.1161/CIR.0000000000001265
3. Mullen M, Jin XY, Child A, et al. Irbesartan in Marfan syndrome (AIMS): a double-blind, placebo-controlled randomised trial. Lancet. 2019;394(10216):2263-2270. doi:10.1016/S0140-6736(19)32518-8
4. Izgi C, Newsome S, Alpendurada F, et al. External Aortic Root Support to Prevent Aortic Dilatation in Patients With Marfan Syndrome. J Am Coll Cardiol. 2018;72(10):1095-1105. doi:10.1016/j.jacc.2018.06.053
5. Kim JH, Baggish AL, Levine BD, et al. Clinical Considerations for Competitive Sports Participation for Athletes With Cardiovascular Abnormalities: A Scientific Statement From the American Heart Association and American College of Cardiology [published correction appears in Circulation. 2025 Apr;151(13):e864. doi: 10.1161/CIR.0000000000001326.]. Circulation. 2025;151(11):e716-e761. doi:10.1161/CIR.0000000000001297
Prognosis for Marfan Syndrome
Advancements in therapy and regular monitoring have reduced mortality for patients with Marfan syndrome, and the expected life span approaches that of the general population (1). However, quality of life is still decreased compared to the general population because of chronic pain, physical limitations, and the psychological burden of the disease.
Neonatal Marfan syndrome has a poor prognosis, with one study showing the majority of patients dying before 16 month of age, and the majority of the survivors requiring cardiac surgery (2).
Prognosis references
1. Dietz H. FBN1-Related Marfan Syndrome. 2001 Apr 18 [Updated 2022 Feb 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025.
2. van der Leest EC, van der Hulst AE, Pals G, et al. Genotype-Phenotype Correlations, Treatment, and Prognosis of Children With Early-Onset (Neonatal) Marfan Syndrome. Clin Genet. 2025;108(2):134-145. doi:10.1111/cge.14722
Key Points
Marfan syndrome results from an autosomal dominant mutation of the FBN1 gene encoding the glycoprotein fibrillin-1, which is the main component of microfibrils, resulting in numerous possible clinical manifestations.
Manifestations vary widely, but the principal structural defects involve the cardiovascular, musculoskeletal, and ocular systems.
Aortic dissection is the most dangerous and life-threatening complication.
Diagnose using clinical criteria, family history, echocardiography, and genetic testing.
Perform cardiac imaging and ophthalmologic evaluation; consider additional imaging.
Give a beta-blocker and/or an angiotensin II receptor blocker to slow aortic dilation and help prevent aortic dissection, offer prophylactic aortic root surgery based on published guidelines, and treat other complications as they arise.Give a beta-blocker and/or an angiotensin II receptor blocker to slow aortic dilation and help prevent aortic dissection, offer prophylactic aortic root surgery based on published guidelines, and treat other complications as they arise.
Drug Information for the Topic





