



Retinitis pigmentosa is a slowly progressive, bilateral degeneration of the retina and retinal pigment epithelium caused by various genetic mutations. Symptoms include night blindness and loss of peripheral vision. Diagnosis is by funduscopy, which shows pigmentation in a bone-spicule configuration in the equatorial retina, narrowing of the retinal arterioles, a waxy pallor of the optic disc, posterior subcapsular cataracts, and cells in the vitreous. Electroretinography helps confirm the diagnosis. Vitamin A palmitate, omega-3 fatty acids, and lutein plus zeaxanthin may help slow progression of vision loss.
Abnormal gene coding for retinal proteins appears to be the cause of retinitis pigmentosa; several genes have been identified. Transmission may be autosomal recessive, autosomal dominant, or, infrequently, X-linked. It may occur as part of a syndrome (eg, abetalipoproteinemia [Bassen-Kornzweig], Laurence-Moon). One of these syndromes includes congenital hearing loss as well (Usher syndrome).
Symptoms and Signs of Retinitis Pigmentosa
Retinal rods are affected, causing defective night vision that becomes symptomatic at varying ages, sometimes in early childhood. Night vision may eventually be lost. A peripheral ring scotoma (detectable by visual field testing) widens gradually, and central vision may also be affected in advanced cases. Vision decreases as the macula becomes increasingly involved and can evolve to legal blindness.
Hyperpigmentation in a bone-spicule configuration in the midperipheral retina is the most conspicuous funduscopic finding. Other findings include the following:
Narrowing of the retinal arterioles
Cystoid macular edema
Waxy yellow appearance of the disc
Posterior subcapsular cataracts
Cells in the vitreous (less common)
Myopia
Diagnosis of Retinitis Pigmentosa
Funduscopy
Electroretinography

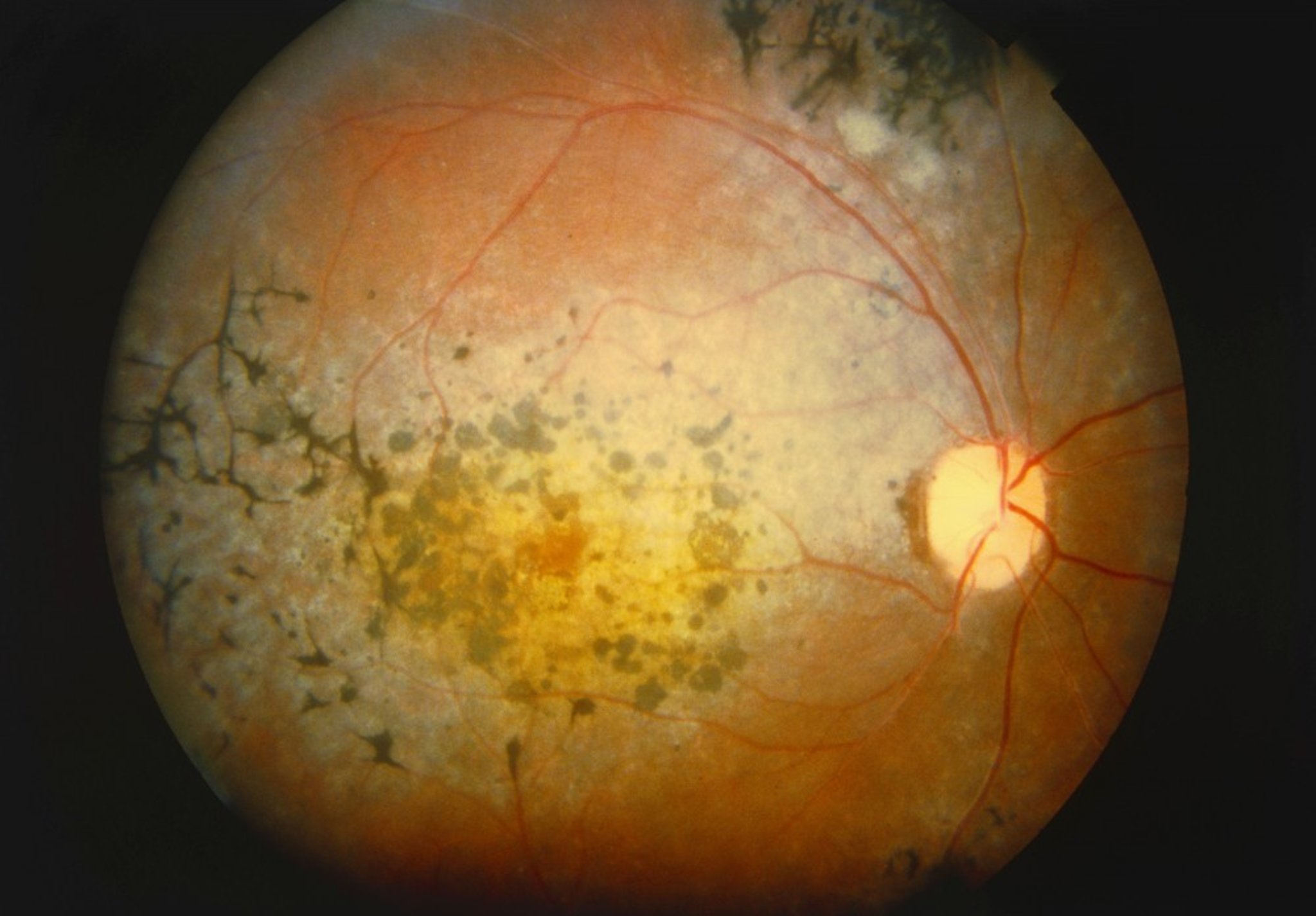
The most characteristic funduscopic finding in retinitis pigmentosa is hyperpigmentation in a bone-spicule configuration. Other findings may include arteriolar narrowing, cystoid macular edema, and a waxy yellow disc surface.
WESTERN OPHTHALMIC HOSPITAL/SCIENCE PHOTO LIBRARY
The diagnosis is suspected in patients with poor night vision or a family history. Diagnosis is by funduscopy, usually supplemented with electroretinography. Other retinopathies that can simulate retinitis pigmentosa should be excluded; they include retinopathies associated with syphilis, rubella, phenothiazine or chloroquine toxicity, and nonocular cancer.
Family members should be examined and tested as necessary or desired to establish the hereditary pattern (1). Patients with a hereditary syndrome may wish to seek genetic counseling before having children.
Diagnosis reference
1. American Academy of Ophthalmology Secretary for Quality of Care; Duncan JL, Branham K, Birch DG, et al. Guidelines on Clinical Assessment of Patients with Inherited Retinal Degenerations-2022. October 2022. Accessed March 19, 2026.
Treatment of Retinitis Pigmentosa
Vitamin A palmitate
Omega-3 fatty acids
Lutein plus zeaxanthin
Carbonic anhydrase inhibitors for cystoid macular edema
Intraocular computer chip implants
There is no way to reverse damage caused by retinitis pigmentosa, but treatment options with gene therapy are emerging (1). Vitamin A palmitate 15,000 IU orally once a day may help slow disease progression in some patients. Patients taking vitamin A palmitate should have regular liver tests. Dietary supplementation with lutein plus zeaxanthin may also slow the rate of vision loss (2).
For patients with cystoid macular edema, carbonic anhydrase inhibitors given orally (eg, acetazolamide) or topically (eg, dorzolamide) may yield mild vision improvement (3). Voretigene neparvovec is available for the treatment of confirmed biallelic RPE65 mutation-associated retinal dystrophy. This is an adenovirus vector-based gene therapy that is injected surgically into the subretinal space in patients who have viable retinal cells and this specific mutation (4). This treatment can restore ambulatory vision in these patients. For patients with total or near total vision loss, epiretinal and subretinal computer chip implants can restore some visual sensations.
Treatment references
1. Nguyen XT, Moekotte L, Plomp AS, Bergen AA, van Genderen MM, Boon CJF. Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. Int J Mol Sci. 2023;24(8):7481. doi:10.3390/ijms24087481
2. Kumar P, Banik SP, Ohia SE, et al. Current insights on the photoprotective mechanism of the macular carotenoids, lutein and zeaxanthin: Safety, efficacy and bio-delivery. J Am Nutr Assoc. 2024;43(6):505-518. doi: 10.1080/27697061.2024.2319090
3. Bakthavatchalam M, Lai FH, Rong SS, et al. Treatment of cystoid macular edema secondary to retinitis pigmentosa: A systematic review. Surv Ophthalmol. 2018;63(3):329–339. doi: 10.1016/j.survophthal.2017.09.009
4. Maguire AM, Russell S, Wellman JA, et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: Results of phase 1 and 3 trials. Ophthalmology. 2019;126(9):1273-1285. doi: 10.1016/j.ophtha.2019.06.017
Key Points
Early symptoms of retinitis pigmentosa include defective night vision and peripheral vision.
Diagnose by hyperpigmentation in a bone-spicule configuration on funduscopy and confirm with electroretinography.
Prescribe vitamin A, lutein, and zeaxanthin supplements to decrease rate of vision loss.
Treat patients with cystoid macular edema with carbonic anhydrase inhibitors.
Gene therapy with voretigene neparvovec can restore ambulatory vision in patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy.
Drug Information for the Topic





