



Hereditary spherocytosis and hereditary elliptocytosis are congenital red blood cell (RBC) membrane disorders that can cause a mild hemolytic anemia. Symptoms, generally milder in hereditary elliptocytosis, include variable degrees of anemia, jaundice, and splenomegaly. Diagnosis requires demonstration of increased RBC osmotic fragility and a negative direct antiglobulin test. Genetic testing is often performed. Rarely, patients < 45 years with symptomatic disease require splenectomy.
Hereditary spherocytosis (chronic familial icterus; congenital hemolytic jaundice; familial spherocytosis; spherocytic anemia) is an autosomal dominant disease with variable gene penetrance due to mutations in spectrin, ankyrin, band 3, or protein 4.2 (1). Approximately 25% of cases are sporadic (de novo dominant mutation) or autosomal recessive (rare) (2). Hereditary spherocytosis is characterized by hemolysis of spheroidal RBCs and anemia.
Hereditary elliptocytosis (ovalocytosis) is a rare autosomal dominant disorder in which RBCs are oval or elliptical due to mutations in spectrin, protein 4.1, or glycophorin C. Hemolysis is usually absent or slight, with little or no anemia except in some patients who are homozygous (hereditary pyropoikilocytosis) (1).
References
1. Kalfa TA. Diagnosis and clinical management of red cell membrane disorders. Hematology Am Soc Hematol Educ Program. 2021;2021(1):331-340. doi:10.1182/hematology.2021000265
2. Miraglia del Giudice E, Nobili B, Francese M, et al. Clinical and molecular evaluation of non-dominant hereditary spherocytosis. Br J Haematol. 2001;112(1):42-47. doi:10.1046/j.1365-2141.2001.02501.x
Pathophysiology
Alterations in membrane proteins cause the RBC abnormalities in both disorders.
In hereditary spherocytosis, the cell membrane surface area is decreased disproportionately to the intracellular content due to loss of proteins associated with the cell membrane. The decreased surface area of the cell impairs the flexibility needed for the cell to traverse the spleen’s microcirculation, causing intrasplenic hemolysis.
In hereditary elliptocytosis, genetic mutations result in weakness of the cytoskeleton of the cell, leading to deformation of the cell. The abnormally shaped RBCs are taken up and destroyed by the spleen.
Symptoms and Signs
In hereditary spherocytosis, symptoms and signs are usually mild. The anemia may be so well compensated that it is not recognized until an intercurrent viral illness, such as parvovirus infection, transiently decreases RBC production, causing an aplastic crisis. These episodes can be self-limited, resolving with resolution of the infection, while others require urgent treatment. Moderate jaundice and symptoms of anemia are present in severe cases. Splenomegaly is almost invariable but only rarely causes abdominal discomfort. Hepatomegaly may be present. Cholelithiasis (pigment stones) is common and may be the presenting symptom. Although usually one or more family members have had symptoms, several generations may be skipped because of variations in the degree of gene penetrance.
In hereditary elliptocytosis, clinical features are similar to those of hereditary spherocytosis but tend to be milder; splenomegaly may be present.
Diagnosis
Peripheral blood smear, RBC fragility assay, direct antiglobulin (Coombs) test, and genetic testing for membranopathies
Hereditary spherocytosis or hereditary elliptocytosis is suspected in patients with unexplained hemolysis (as suggested by the presence of anemia and reticulocytosis), particularly if splenomegaly, a family history of similar manifestations, or suggestive RBC indices are present.
In hereditary spherocytosis, because RBCs are spheroidal and the mean corpuscular volume (MCV) is normal, the mean corpuscular diameter is below normal, and RBCs resemble spherocytes. The mean corpuscular hemoglobin concentration (MCHC) is increased. Reticulocytosis is common.
In hereditary elliptocytosis, the RBCs are typically elliptical or cigar-shaped; however, the clinical presentation is variable. Diagnosis is usually made by the presence of at least 60% elliptocytes on peripheral smear and a family history of similar disease.

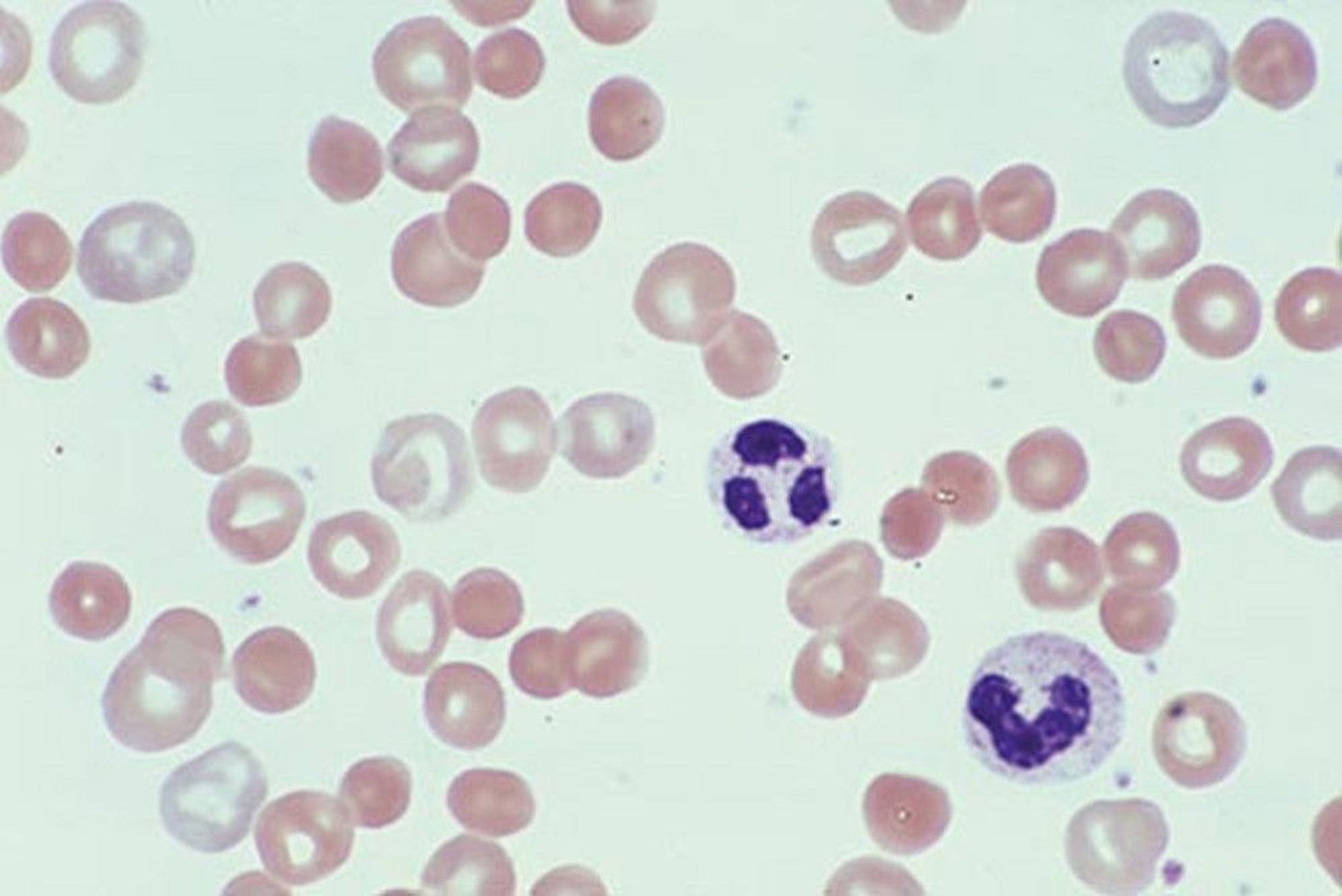
The presence of abundant spherocytes in the peripheral blood smear suggests either hereditary spherocytosis (HS) or autoimmune hemolytic anemia (AIHA). Spherocytes are spherical red blood cells without an area of central pallor and are usually slightly smaller in size than the average red cell. Spherocytes have increased osmotic fragility (because of decreased distensibility associated with reduced surface membrane area) in hypotonic saline, and this test (osmotic fragility test) is positive in HS and in AIHA. The direct antiglobulin (Coombs) test distinguishes between them; the result is positive in AIHA and negative in HS.
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
If either of these disorders is suspected, the following tests are done:
RBC osmotic fragility test, which exposes RBCs to varying concentrations of saline
Other specialized testing for RBC membrane abnormalities including reduced eosin-5-maleimide (EMA) binding and ektacytometry, where available
Direct antiglobulin (direct Coombs) test to exclude spherocytosis due to autoimmune hemolytic anemia
RBC fragility is characteristically increased, but in mild cases of either hereditary spherocytosis or hereditary elliptocytosis, it may be normal unless sterile defibrinated blood is first incubated at 37° C for 24 hours to deplete red cell ATP (adenosine triphosphate) stores. The direct antiglobulin test results are negative.
Following initial evaluation, genetic testing can be performed to confirm the diagnosis and should especially be considered if splenectomy is contemplated; specific panels to test for RBC membranopathies are available.
Treatment
Sometimes splenectomy
Splenectomy, after appropriate vaccination, is the only specific treatment for hereditary spherocytosis or hereditary elliptocytosis. It is indicated in patients with symptomatic hemolysis or complications such as biliary colic or persistent aplastic crisis. If the gallbladder has stones or other evidence of cholestasis, it should be removed.
Although spherocytosis persists after splenectomy, the cells survive longer in the circulation. Usually, symptoms resolve and anemia and reticulocytosis decrease. However, RBC fragility remains high.





