



Coccidioidomycosis is caused by the fungus Coccidioides immitis or C. posadasii; it usually occurs as an acute, benign, asymptomatic or self-limited respiratory infection. The spectrum of disease ranges from acute pneumonia to disseminated extrapulmonary disease (including meningitis). Symptoms, if present, are those of lower respiratory infection or low-grade nonspecific disseminated disease. Diagnosis is suspected based on clinical and epidemiologic characteristics and confirmed by chest radiograph, culture, and serologic testing. Treatment, if needed, is usually with fluconazole, itraconazole, and other triazole antifungals, or amphotericin B.
Coccidioidomycosis is a systemic fungal infection caused by inhalation of airborne arthroconidia (spores) of the dimorphic fungi Coccidioides immitis and C. posadasii.
In North America, the endemic area for coccidioidomycosis includes:
The southwestern United States
Northern Mexico
The affected areas are arid or semiarid regions of the southwestern United States including Arizona, the central valley of California, parts of New Mexico, and Texas west of El Paso. The area extends into northern Mexico, and foci occur in parts of Central America and Argentina. Coccidioidomycosis also occurs in Utah, Nevada, and southcentral Washington. Approximately 20,000 cases are reported to the Centers for Disease Control and Prevention (CDC) annually (1).
Coccidioidomycosis causes an estimated 15 to 30% of cases of community-acquired pneumonia in highly endemic areas of Arizona such as the metropolitan areas of Tucson and Phoenix.
(See also Overview of Fungal Infections.)
General reference
1. Centers for Disease Control and Prevention (CDC): Reported Cases of Valley Fever (Coccidioidomycosis). Accessed July 22, 2025.
Pathophysiology of Coccidioidomycosis
Coccidioidomycosis is acquired by inhaling spores. Spores are present in soil and can become airborne in dust that can travel downwind. Thus, certain occupations (eg, farming, construction) and outdoor recreational activities are associated with an increased risk of infection. Epidemics can occur when heavy rains, which promote the growth of mycelia, are followed by drought and winds. Because of travel and the delayed onset of clinical manifestations, infections can become evident outside endemic areas.
Once inhaled, Coccidioides spores convert to large tissue-invasive spherules. As spherules enlarge and then rupture, each releases thousands of small endospores, which may form new spherules.
Pulmonary disease is characterized by an acute, subacute, or chronic granulomatous reaction with varying degrees of fibrosis. Lesions may cavitate or form nodular, coin-like lesions.
Sometimes disease progresses, with widespread lung involvement, systemic dissemination, or both; focal lesions may form in almost any tissue, most commonly in skin, subcutaneous tissues, bones (osteomyelitis), joints, and meninges (meningitis).
Risk factors for progressive coccidioidomycosis
Progressive coccidioidomycosis is uncommon in otherwise healthy people and more likely to occur in the following contexts:
HIV infection
Use of immunosuppressants
Advanced age
Second or third trimester of pregnancy or postpartum
People who are Filipino, Black, American Indian, Hispanic, or Asian (in decreasing order of relative risk)
Cancer
After stem cell or solid organ transplantation
Symptoms and Signs of Coccidioidomycosis
Primary coccidioidomycosis
Most patients with primary coccidioidomycosis are asymptomatic, but nonspecific respiratory symptoms resembling those of influenza, acute bronchitis, or, less often, acute pneumonia (ie, community-acquired pneumonia) or pleural effusion sometimes occurs 1 to 3 weeks after initial infection.
Symptoms of primary coccidioidomycosis, in decreasing order of frequency, include fever, cough, chest pain, chills, sputum production, sore throat, and hemoptysis.
Physical signs may be absent or limited to scattered rales with or without areas of dullness to percussion over lung fields. Some patients can develop hypersensitivity to the localized respiratory infection such as a serum sickness–like reaction (also called Valley fever triad), manifested by arthritis, conjunctivitis, and a rash (either erythema nodosum or erythema multiforme).

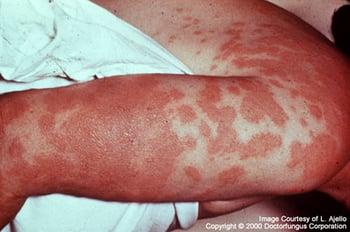
Hypersensitivity to Coccidioides immitis antigens may manifest as erythema nodosum, arthritis, conjunctivitis, or erythema multiforme. This photo is an example of erythema multiforme due to coccidioidomycosis.
Primary pulmonary lesions sometimes leave nodular coin-shaped lesions that must be distinguished from tumors, tuberculosis, and other granulomatous infections. Sometimes residual cavitary lesions develop; they may vary in size over time and often appear thin-walled. A small percentage of these cavities fail to close spontaneously. Hemoptysis or the threat of rupture into the pleural space occasionally necessitates surgery.
Progressive coccidioidomycosis
Nonspecific symptoms develop a few weeks, months, or occasionally years after primary infection; they include low-grade fever, anorexia, weight loss, and weakness.
Cutaneous manifestations are due to immunologically induced reactive eruptions, dissemination of the organisms from the lungs, or direct inoculation (primary cutaneous infection). Erythema nodosum is the most frequent reactive eruption associated with coccidioidomycosis and is characterized by multiple, self-limited, erythematous, painful, subcutaneous nodules usually on the lower extremities that appear 1 to 3 weeks after the initial respiratory symptoms. A generalized toxic exanthem and erythema multiforme have also been reported. The Valley fever triad (conjunctivitis, arthritis, and rash), a hypersensitivity reaction, may occur with progressive disease.
Extensive pulmonary involvement is uncommon in otherwise healthy people and occurs mainly in those who are immunocompromised. It may cause progressive cyanosis, dyspnea, and mucopurulent or bloody sputum.
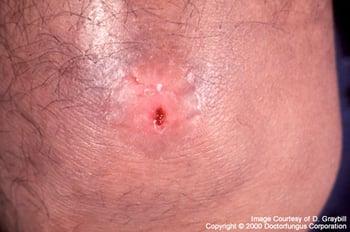
Coccidioidomycosis that disseminates from a primary pulmonary infection may manifest as a single skin lesion.
Symptoms of extrapulmonary lesions depend on the site. Draining sinus tracts sometimes connect deeper lesions to the skin. Localized extrapulmonary lesions often become chronic and recur frequently, sometimes long after completion of seemingly successful antifungal therapy.
Untreated disseminated coccidioidomycosis is usually fatal and, if meningitis is present, is uniformly fatal without prolonged and possibly lifelong treatment. Case fatality rates in patients with advanced HIV infection are approximately 70% with a median survival of 54 days (1).
Symptoms and signs reference
1. Singh VR, Smith DK, Lawerence J, et al. Coccidioidomycosis in patients infected with human immunodeficiency virus: review of 91 cases at a single institution. Clin Infect Dis. 1996;23(3):563-568. doi:10.1093/clinids/23.3.563
Diagnosis of Coccidioidomycosis
Cultures (routine or fungal)
Microscopic examination of specimens to check for Coccidioides spherules
Imaging studies (chest radiographs or CT scans)
Serologic testing
Molecular diagnostics
The diagnosis of coccidioidomycosis is suspected based on history and typical physical findings, when apparent. Confirmation can be established by fungal culture or by visualization of Coccidioides spherules in sputum, pleural fluid, cerebrospinal fluid (CSF), exudate from draining lesions, or histopathologic specimens obtained by biopsy (1). Intact spherules are usually 20 to 80 micrometers in diameter, thick-walled, and filled with small (2 to 4 micrometers) endospores. Endospores released into tissues from ruptured spherules may be mistaken for nonbudding yeasts. Because culturing Coccidioides can pose a severe biohazard to laboratory personnel, the laboratory should be notified of the suspected diagnosis.
Chest imaging findings can be helpful adjuncts for diagnosis.
Serologic testing for anticoccidioidal antibodies includes (2):
Enzyme immunoassay, which is very sensitive and is commonly used to diagnose coccidioidomycosis
Immunodiffusion (to detect IgM or IgG antibodies)
Complement fixation (to detect IgG antibodies)
Titers ≥ 1:4 in serum are consistent with current or recent infection, and high titers (≥ 1:32) signify an increased likelihood of extrapulmonary dissemination. Complement fixation titers can be used to estimate disease severity (2); high titers suggest more severe disease. However, immunocompromised patients may have low titers. Titers normally decline during successful therapy.
The presence of complement-fixing antibodies in CSF is diagnostic of coccidioidal meningitis and is preferred to CSF cultures because they are rarely positive.
A urine antigen test may be useful for diagnosing coccidioidomycosis in immunocompromised patients with severe forms of the disease, including pneumonia and disseminated infection.
Serum eosinophilia may be an important clue in identifying coccidioidomycosis.
DNA probes can rapidly identify the fungus once growth occurs in the laboratory. Using PCR techniques to test lower respiratory tract samples for DNA can provide a more rapid diagnosis. However, this test is not widely available.
Diagnosis references
1. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. doi:10.1093/cid/ciw360
2. Miller JM, Binnicker MJ, Campbell S, et al. Guide to Utilization of the Microbiology Laboratory for Diagnosis of Infectious Diseases: 2024 Update by the Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM). Clin Infect Dis. Published online March 5, 2024. doi:10.1093/cid/ciae104
Treatment of Coccidioidomycosis
For mild to moderate disease, fluconazole or itraconazole
For severe disease, amphotericin B
Rarely surgery
(See also Antifungal Medications.)
Patients with primary coccidioidomycosis and risk factors for severe or progressive disease should be treated.
Most immunocompetent patients with mild initial pulmonary infection do not require antifungal therapy, especially if asymptomatic, but oral fluconazole for 3 to 6 months may be considered if symptoms are prolonged or severe (1). Symptoms may resolve more quickly in treated patients than in those who are not treated with an antifungal. However, fluconazole may blunt the immune response, and the risk of hematogenous seeding in primary infection may not be high enough to warrant the use of it; both factors should be considered before initiating treatment. High complement fixation titers indicate spread and the need for treatment.
Mild to moderate nonmeningeal extrapulmonary involvement should be treated with fluconazole or itraconazole. Voriconazole or posaconazole delayed-release tablets are alternatives but have not been well-studied.
Treatment is typically extended for a period of 1 or 2 years in patients with skin, soft-tissue, or bone involvement. All patients with bone and/or joint infection should receive antifungal therapy with a triazole. Itraconazole may be more effective than fluconazole, but it requires monitoring of therapeutic levels and carries a slightly higher risk of adverse events and drug interactions (2).
For severe illness, lipid formulations of amphotericin B are preferred over conventional amphotericin B. Patients can usually be switched to an oral triazole once they have been stabilized, usually within several weeks.
Immunocompromised patients with coccidioidomycosis require maintenance therapy to prevent relapse; oral fluconazole or oral itraconazole usually is sufficient and is administered until the CD4 cell count becomes > 250 cells/mcL on highly active antiretroviral therapy (HAART).
In pregnant patients, triazole antifungals should be avoided in the first trimester because of the risk of teratogenicity. Women with mild to moderate disease in the first trimester typically do not require treatment. For women with severe or extrapulmonary disease in the first trimester, treatment with amphotericin B is recommended. After the first trimester, a triazole antifungal can be considered. Patients who acquire infection in the second or third trimester or within 6 weeks postpartum are at increased risk of disease progression. If not treated, they should be monitored clinically by checking serial complement fixation titers.
For meningeal coccidioidomycosis, fluconazole is used. The optimal dose is unclear; a high oral dose once a day may be more effective. Patients should continue azole maintenance therapy for life because relapses are common and potentially fatal.
Surgical removal of involved bone may be necessary to cure osteomyelitis.
When residual cavitary pulmonary lesions cause hemoptysis or are likely to rupture, surgery may be necessary.
Treatment references
1. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. doi:10.1093/cid/ciw360
2. Galgiani JN, Catanzaro A, Cloud GA, et al. Comparison of oral fluconazole and itraconazole for progressive, nonmeningeal coccidioidomycosis. A randomized, double-blind trial. Mycoses Study Group. Ann Intern Med. 2000;133(9):676–686. doi:10.7326/0003-4819-133-9-200011070-00009
Key Points
Coccidioidomycosis is a common fungal infection in endemic areas and is acquired by inhaling spore-laden dust.
It is endemic to the southwestern United States and northern Mexico; disease also occurs in certain parts of Central and South America.
Most patients have an asymptomatic or mild pulmonary infection, but those who are immunocompromised or have other risk factors may develop severe, progressive pulmonary disease or disseminated infection (typically to skin and soft tissue, bone, or meninges).
The diagnosis is based on culture, staining, and/or serologic testing.
For mild to moderate disease, use fluconazole or itraconazole.
For severe disease, use a lipid formulation of amphotericin B.
Drug Information for the Topic





