


A neuron generates and propagates an action potential along its axon, then transmits this signal across a synapse by releasing neurotransmitters, which trigger a reaction in another neuron or an effector cell (eg, muscle cells, most exocrine and endocrine cells). Neurotransmitters enable neurons to communicate with each other. Neurotransmitters that are released bind to receptors on another neuron. Neurons that release neurotransmitters are called presynaptic neurons. Neurons that receive neurotransmitter signals are called postsynaptic neurons. The signal may stimulate or inhibit the receiving cell, depending on the neurotransmitter and receptor involved. Other factors, including drugs and disorders, affect communication between neurons by modulating the production and actions of neurotransmitters including
Their release, reuptake, and degradation
The number and function of postsynaptic neurotransmitter receptors
Sometimes signals between neurons occur in the reverse direction (called retrograde neurotransmission). In such cases, the dendrites (a neuron’s receiving branches) on the postsynaptic neurons release neurotransmitters that affect receptors on the presynaptic neurons. Retrograde transmission can inhibit presynaptic neurons from releasing additional neurotransmitters and help control the level of activity and communication among neurons.
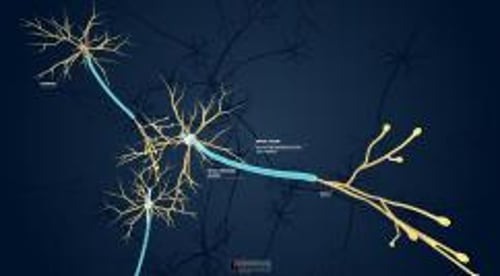
In the central nervous system (CNS), interconnections are complex. An impulse from one neuron to another may pass from
Axon to cell body
Axon to dendrite
Cell body to cell body
Dendrite to dendrite
A neuron can simultaneously receive many impulses—excitatory and inhibitory—from other neurons and integrate simultaneous impulses into various patterns of firing.
Propagation
Action potential propagation along an axon is electrical, caused by the exchanges of sodium and potassium ions across the axonal membrane. A particular neuron generates the same action potential after each stimulus, conducting it at a fixed velocity along the axon. Velocity depends on axonal diameter and degree of myelination and ranges from 1 to 4 m/sec in small unmyelinated fibers to 75 m/sec in large myelinated ones. Propagation speed is higher in myelinated fibers because the myelin cover has regular gaps (nodes of Ranvier) where the axon is exposed. The electrical impulse jumps from one node to the next, skipping the myelinated section of the axon. Thus, disorders that alter the myelin cover (eg, multiple sclerosis, Guillain-Barré syndrome) interfere with impulse propagation, causing various neurologic symptoms.
Transmission
Impulse transmission is chemical, caused by release of specific neurotransmitters from the nerve ending (terminal). Neurotransmitters diffuse across the synaptic cleft and bind briefly to specific receptors on the adjoining neuron or effector cell. Depending on the receptor, the response may be excitatory or inhibitory. Usually, neurons do not touch each other; instead, they communicate through the transmission of neurotransmitters across the synapses. Under some conditions, neurons near each other can communicate using electrical impulses across a gap junction..
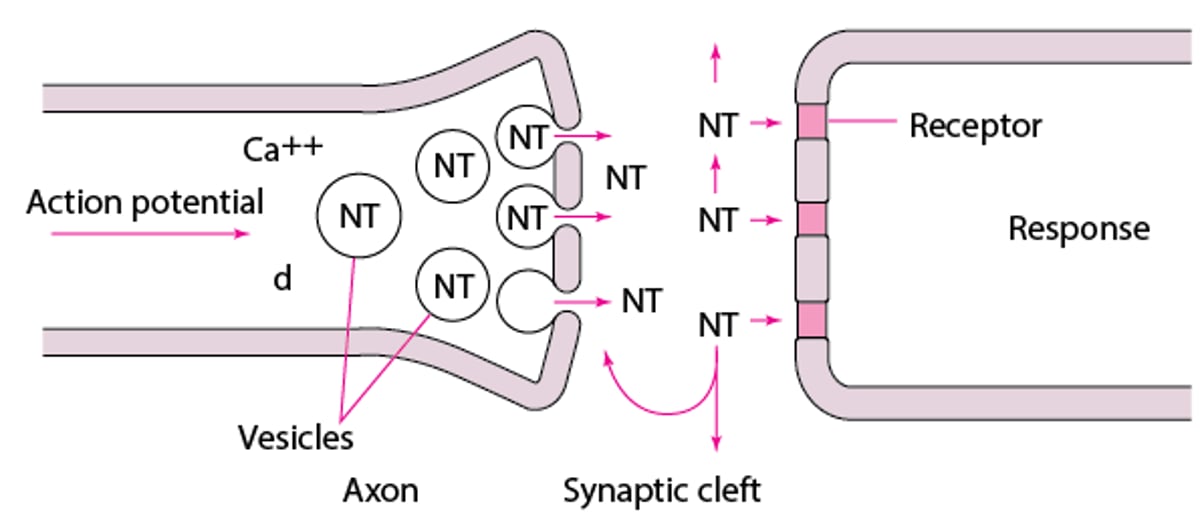
The nerve cell body produces enzymes that synthesize most neurotransmitters, which are stored in vesicles at the nerve terminal (see figure Neurotransmission). The amount in one vesicle (usually several thousand molecules) is a quantum. A membrane action potential arriving at the terminal opens axonal calcium channels; calcium inflow releases neurotransmitter molecules from many vesicles by fusing the vesicle membranes to the nerve terminal membrane. Membrane fusion generates an opening through which the molecules are expelled into the synaptic cleft via exocytosis.
One type of synapse, the electrical synapse, does not involve neurotransmitters; ion channels directly connect the cytoplasm of the presynaptic and postsynaptic neurons. This type of transmission is the fastest.
Excitatory and inhibitory signals
The reaction triggered by neurotransmitter release can either excite or activate the postsynaptic neuron or inhibit or block its activity. Postsynaptic neurons receive multiple neurotransmitter signals and electrical signals from many neurons. The receiving neuron ultimately adds the inputs together, and if more excitatory signals are received, the neuron fires and sends signals to other neurons. If the sum of the signals are inhibitory, the neuron does not fire and does not influence the activity of other neurons. This adding up of responses is called summation. Neurotransmitters thus facilitate rapid communication between neurons by changing the firing of the action potential.
Other forms of summation include
Spatial summation: When multiple impulses are received on different locations of the neuron and the neuron then sums them up
Temporal summation: When impulses are received within a short period of time and are then added together
For a neuron to generate a signal and fire, it must reach a threshold potential. A threshold potential is produced by a net increase in sodium influx into the cell during the exchange of sodium and potassium ions. When enough sodium enters the cell, the threshold is reached; when the threshold is reached, an action potential is fired; it travels along the neuron's membrane. The threshold must be reached for an action potential to be generated.
Neurotransmission
Action potentials open the axonal calcium (Ca) channels (not shown). Ca++ activates release of neurotransmitters (NT) from vesicles where they are stored. NT molecules fill the synaptic cleft. Some bind to postsynaptic receptors, initiating a response. The others are pumped back into the axon and stored or diffuse into the surrounding tissues. |

The amount of neurotransmitters in the terminal is typically independent of nerve activity and kept relatively constant by modifying uptake of neurotransmitter precursors or the activity of enzymes involved in neurotransmitter synthesis or destruction. Stimulation of presynaptic receptors can decrease presynaptic neurotransmitter synthesis, and blockade can increase it.
The neurotransmitter-receptor interaction must be terminated quickly to end the continued action of the neurotransmitter and/or to allow rapid, repeated activation of receptors. One of the following can happen to neurotransmitters that have interacted with receptors:
They can be quickly pumped back into the presynaptic nerve terminals by active, ATP-dependent processes (reuptake) for recycling or destruction.
They can be eliminated by enzymes near the receptors.
They can diffuse into the surrounding area and be removed.
Neurotransmitters taken up by the nerve terminals are repackaged in granules or vesicles in the axon terminal for reuse.
Some types of single neurons can release two or more different neurotransmitters (called cotransmission)—for example, acetylcholine and glutamate. Multiple neurotransmitters may act on a single postsynaptic neuron or affect multiple postsynaptic neurons. Cotransmission allows for intricate communication among neurons to control different events in the CNS and the peripheral nervous system (PNS).
Neurotransmitters can also facilitate more long-term changes that involve additional pathways such as changes in the activity of genes and proteins.
Receptors
Neurotransmitter receptors are protein complexes that span the cell membrane. Their nature determines whether a given neurotransmitter is excitatory or inhibitory. Receptors that are continuously stimulated by neurotransmitters or drugs become desensitized (downregulated); those that are not stimulated by their neurotransmitter or are chronically blocked by drugs become supersensitive (upregulated). Downregulation or upregulation of receptors strongly influences the development of tolerance and physical dependence. These concepts are particularly important in organ or tissue transplantation, in which denervation deprives receptors of their neurotransmitter; as a result, transplanted organs may become overly sensitive to neural stimulation. Withdrawal symptoms can be explained at least in part by a rebound phenomenon due to altered receptor affinity or density.
Most neurotransmitters interact primarily with postsynaptic receptors, but some receptors are located on presynaptic neurons, providing fine control of neurotransmitter release.
One family of receptors, termed ionotropic receptors (eg, N-methyl-d
Major Neurotransmitters and Receptors
At least 100 substances can act as neurotransmitters; about 18 are of major importance. Several occur in slightly different forms. Neurotransmitters can be grouped in different classes, such as
adenosine, acetylcholine, serotonin, histamine, noradrenaline)
Neuropeptides (eg, endorphins)
Gaseous molecules (eg, nitric oxide, carbon monoxide)
Endocannabinoids
Glutamate and aspartate
These amino acids (glutamate and aspartate) are the major excitatory neurotransmitters in the CNS. They occur in the cortex, cerebellum, and spinal cord. In neurons, synthesis of nitric oxide (NO) increases in response to glutamate. Excess glutamate can be toxic, increasing intracellular calcium, free radicals, and proteinase activity. These neurotransmitters may contribute to tolerance to opioid therapy and mediate hyperalgesia.
Glutamate receptors (stimulated by glutamate and less strongly by aspartate) are classified as NMDA (N-methyl-d
Gamma-aminobutyric acid
Serotonin
Acetylcholine
Acetylcholine is the major neurotransmitter of the bulbospinal motor neurons, autonomic preganglionic fibers, postganglionic cholinergic (parasympathetic) fibers, and many neurons in the CNS (eg, basal ganglia, motor cortex). It is synthesized from choline and acetyl coenzyme A by choline acetyltransferase, and its action is rapidly terminated via local hydrolysis to choline and acetate by acetylcholinesterase. Acetylcholine levels are regulated by choline acetyltransferase and by choline uptake. Levels of this neurotransmitter are decreased in patients with Alzheimer disease.
Cholinergic receptors are classified as nicotinic N1 (in skeletal muscle and the neuromuscular junction) or N2 (in the central and peripheral nervous systems, including the parasympathetic and sympathetic nervous systems and the adrenal medulla) or muscarinic M1 through M5 (widely distributed in the CNS). M1 occurs in the autonomic nervous system, striatum, cortex, and hippocampus; M2 occurs in the autonomic nervous system, heart, intestinal smooth muscle, hindbrain, and cerebellum.
Dopamine
Dopamine interacts with receptors on some peripheral nerve fibers and many central neurons (eg, in the substantia nigra, midbrain, ventral tegmental area, and hypothalamus). The amino acid tyrosine is taken up by dopaminergic neurons and converted by tyrosine hydroxylase to 3,4-dihydroxyphenylalanine (dopa), which is decarboxylated by aromatic-l-amino-acid decarboxylase to dopamine. After release and interaction with receptors, dopamine is actively pumped back (reuptake) into the nerve terminal. Tyrosine hydroxylase and MAO (which breaks down dopamine) regulate dopamine levels in nerve terminals.
Norepinephrine
Norepinephrine is the neurotransmitter of most postganglionic sympathetic fibers and many central neurons (eg, in the locus caeruleus and hypothalamus). The precursor tyrosine is converted to dopamine, which is hydroxylated by dopamine beta-hydroxylase to norepinephrine. After release and interaction with receptors, some norepinephrine is degraded by catechol O-methyltransferase (COMT), and the remainder is actively taken back into the nerve terminal, where it is degraded by MAO. Tyrosine hydroxylase, dopamine beta-hydroxylase, and MAO regulate intraneuronal norepinephrine levels.
Adrenergic receptors are classified as alpha-1 (postsynaptic in the sympathetic system), alpha-2 (presynaptic in the sympathetic system and postsynaptic in the brain), beta-1 (in the heart), or beta-2 (in other sympathetically innervated structures).
Endorphins and enkephalins
Endorphins and enkephalins are opioids.
Endorphins are large polypeptides that activate many central neurons (eg, in the hypothalamus, amygdala, thalamus, and locus caeruleus). The cell body contains a large polypeptide called pro-opiomelanocortin, the precursor of alpha-, beta-, and gamma-endorphins. Pro-opiomelanocortin is transported down the axon and cleaved into fragments; one is beta-endorphin, contained in neurons that project to the periaqueductal gray matter, limbic structures, and major catecholamine-containing neurons in the brain. After release and interaction with receptors, beta-endorphin is hydrolyzed by peptidases.
Enkephalins include met-enkephalin and leu-enkephalin, which are small polypeptides present in many central neurons (eg, in the globus pallidus, thalamus, caudate, and central gray matter). Their precursor, proenkephalin, is formed in the cell body, then split by specific peptidases into the active peptides. These substances are also localized in the spinal cord, where they modulate pain signals. The neurotransmitters of pain signals in the posterior horn of the spinal cord are glutamate and substance P. Enkephalins decrease the amount of neurotransmitter released and hyperpolarize (make more negative) the postsynaptic membrane, reducing the generation of action potentials and pain perception at the level of the postcentral gyrus. After release and interaction with peptidergic receptors, enkephalins are hydrolyzed into smaller, inactive peptides and amino acids. Rapid inactivation of exogenous enkephalins prevents these substances from being clinically useful. More stable molecules (eg, morphine) are used as analgesics instead.
Endorphin-enkephalin (opioid) receptors are classified as mu-1 and mu-2 (affecting sensorimotor integration and analgesia), delta-1 and delta-2 (affecting motor integration, cognitive function, and analgesia), and kappa-1, kappa-2, and kappa-3 (affecting water balance regulation, analgesia, and food intake). Sigma receptors, currently classified as nonopioid and mostly localized in the hippocampus, bind PCP. New data suggest the presence of many more receptor subtypes, with pharmacologic implications. Components of the molecular precursor to the receptor protein can be rearranged during receptor synthesis to produce several receptor variants (eg, 27 splice variants of the mu opioid receptor). Also, two receptors can combine (dimerize) to form a new receptor.
Other neurotransmitters
Dynorphins are a group of 7 peptides with similar amino acid sequences. They, like enkephalins, are opioids.
Substance P, a peptide, occurs in central neurons (in the habenula, substantia nigra, basal ganglia, medulla, and hypothalamus) and is highly concentrated in the dorsal root ganglia. Its release is triggered by intense afferent painful stimuli. It modulates the neural response to pain and mood; it modulates nausea and vomiting through the activation of NK1A receptors that are localized in the brain stem.
Nitric oxide (NO) is a labile gas that mediates many neuronal processes. It is generated from arginine by NO synthase. Neurotransmitters that increase intracellular calcium (eg, substance P, glutamate, acetylcholine) stimulate NO synthesis in neurons that express NO synthetase. NO may be an intracellular messenger; it may diffuse out of a cell into a second neuron and produce physiologic responses (eg, long-term potentiation [strengthening of certain presynaptic and postsynaptic responses—a form of learning]) or enhance glutamate (NMDA-receptor–mediated) neurotoxicity (eg, in Parkinson disease, stroke, or Alzheimer disease). NO affects other neurotransmitters (eg, GABA and acetylcholine) by changing the influx of calcium into cells to increase the release of other neurotransmitters.
Additional gaseous neurotransmitters include carbon monoxide (CO) and hydrogen sulfide (H2S). These transmitters are produced in cells throughout the body (including the brain). Endogenous CO is generated from the metabolism of heme and can participate in processes that involve fever generation, inflammation, cell survival, and control of blood vessel dilation. Several enzymes are involved in the production of H2S, which is believed to be necessary for the brain to form and retain memories.
Substances with less firmly established roles in neurotransmission include histamine, vasopressin, vasoactive intestinal peptide, carnosine, bradykinin, cholecystokinin, bombesin, somatostatin, corticotropin-releasing factor, neurotensin, and possibly adenosine.
Endocannabinoids are endogenous lipid-based neurotransmitters that modulate brain, endocrine, and immune system function.
Disorders Associated With Defects in Neurotransmission
Disorders or substances that alter the production, release, reception, breakdown, or reuptake of neurotransmitters or that change the number and affinity of receptors can cause neurologic or psychiatric symptoms and cause disease (see table Examples of Disorders Associated With Defects in Neurotransmission). Drugs that modify neurotransmission can alleviate many of these disorders (eg, Parkinson disease, depression).




