



The urinary tract is a common location for congenital anomalies of varying significance. Many anomalies are asymptomatic and diagnosed via prenatal ultrasound or part of a routine evaluation for other congenital anomalies. Other anomalies are diagnosed during evaluation of urinary tract obstruction, urinary tract infection, or trauma.
(See also Congenital Renal Cystic Dysplasia.)
Autosomal Recessive Polycystic Kidney Disease
Incidence of autosomal recessive polycystic kidney disease (ARPKD) is approximately 1/10,000 to 1/20,000 births (1); it is caused by a mutation in the PKHD1 gene, located in chromosome 6p21. In contrast, autosomal dominant polycystic kidney disease is much more common, occurring in about 1/400 to 1/1000 live births (2).
Symptoms of autosomal dominant polycystic kidney disease are usually not present until adulthood. Rarely, symptoms manifest in infancy in the more aggressive form. Children may be diagnosed earlier when a cyst is found incidentally or via family history.
In autosomal recessive polycystic kidney disease, the kidneys and liver are affected. The kidneys are usually greatly enlarged and contain small cysts; renal failure is common in childhood. The liver is enlarged and has periportal fibrosis, bile duct proliferation, and scattered cysts; the remainder of the hepatic parenchyma is normal. Fibrosis causes portal hypertension by age 5 to 10 years, but hepatic function is normal or minimally impaired.
Disease severity and progression vary. Severe disease may be detected prenatally, or soon after birth or in early childhood with renal-related symptoms; less severely affected patients present in late childhood or adolescence with hepatic-related symptoms.
Affected neonates have a protuberant abdomen with huge, firm, smooth, symmetric kidneys. Severely affected neonates commonly have pulmonary hypoplasia secondary to the in utero effects of renal dysfunction and oligohydramnios.
In patients aged 5 to 10 years, signs of portal hypertension, such as esophageal and gastric varices and hypersplenism, occur. If the patient presents in adolescence, nephromegaly is less marked, renal insufficiency may be mild to moderate, and the major symptoms are those related to portal hypertension.
Diagnosis of autosomal recessive polycystic kidney disease may be difficult, especially without a family history. Ultrasound may show renal or hepatic cysts; definitive diagnosis may require biopsy. Ultrasound in late pregnancy usually allows presumptive in utero diagnosis. If postnatal ultrasound is not definitive, MRI or CT may be diagnostic. If needed, molecular testing for PKHD1 can be done when clinical criteria are not met.
Many neonates die in the first few days or weeks of life of pulmonary insufficiency. Most who survive develop progressive renal failure often requiring renal replacement therapy. Experience with renal transplantation with or without hepatic transplantation is limited. When transplantation is done, hypersplenism must be controlled to obviate difficulty with hypersplenism-induced leukopenia, which increases the risk of systemic infection. Portal hypertension may be treated by portacaval or splenorenal shunts, which reduce morbidity but not mortality.
ARPKD references
1. National Institute of Diabetes and Digestive and Kidney Diseases: Autosomal Recessive Polycystic Kidney Disease. Accessed July 31, 2024.
2. National Organization for Rare Disorders: Autosomal Dominant Polycystic Kidney Disease. Accessed August 6, 2024.
Duplication Anomalies
Supernumerary collecting systems may be unilateral or bilateral and may involve the renal pelvis and ureters (accessory renal pelvis, double or triple pelvis and ureter), calyx, or ureteral orifice. Duplex kidneys have a single renal unit with more than 1 collecting system. This anomaly differs from fused kidneys, which involves fusion of 2 renal parenchymal units maintaining their respective individual collecting systems. Some duplication anomalies have ureteral ectopy with or without ureterocele and/or vesicoureteral reflux (VUR).
Management depends on the anatomy and function of each separately drained segment. Surgery may be necessary to correct obstruction or VUR.
Fusion Anomalies
With fusion anomalies, the kidneys are joined, but the ureters enter the bladder on each side. These anomalies increase the risk of ureteropelvic junction obstruction, vesicoureteral reflux, congenital renal cystic dysplasia, and injury caused by anterior abdominal trauma.
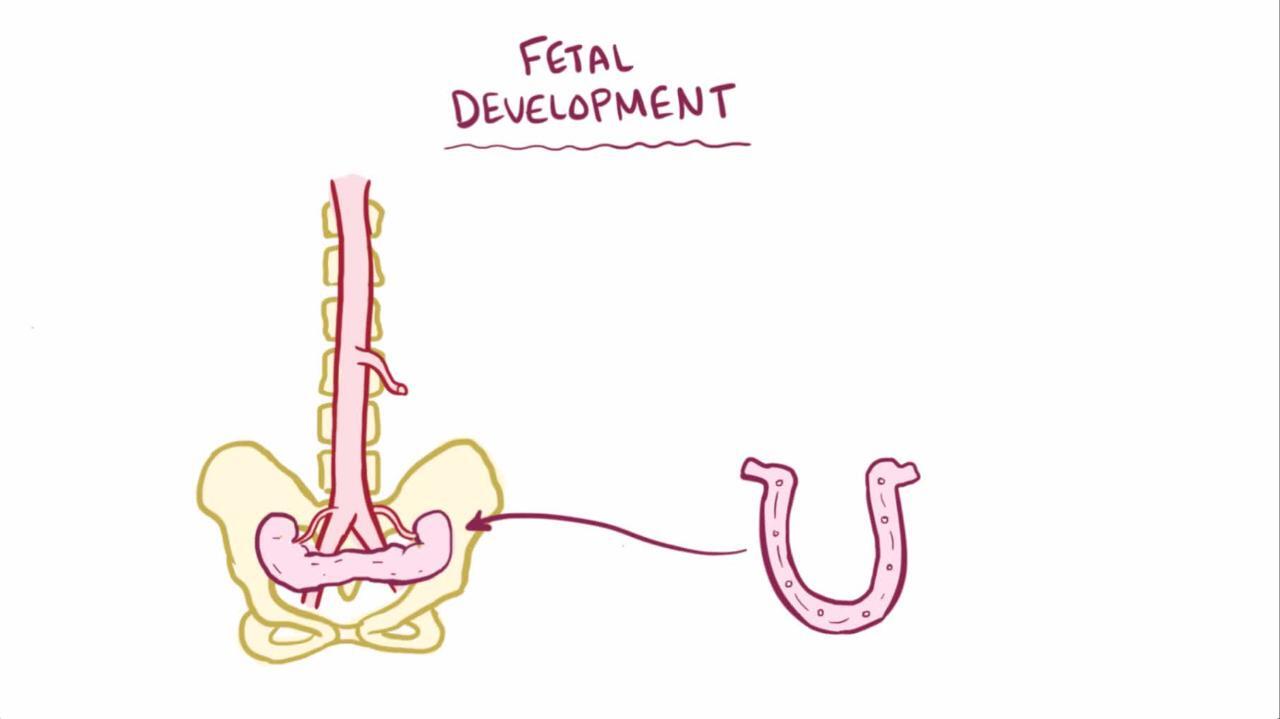
Horseshoe kidney
Horseshoe kidney, the most common fusion anomaly, occurs when renal parenchyma on each side of the vertebral column is joined at the corresponding (usually lower) poles; an isthmus of renal parenchyma or fibrous tissue joins at the midline. The ureters course medially and anteriorly over this isthmus and generally drain well. Obstruction, if present, is usually secondary to insertion of the ureters high in the renal pelvis.
Pyeloplasty relieves the obstruction and can be done without resecting the isthmus.

Crossed fused renal ectopia
Crossed fused renal ectopia is the second most common fusion anomaly. The renal parenchyma (representing both kidneys) is on one side of the vertebral column. One of the ureters crosses the midline and enters the bladder on the side opposite the fused kidneys.
When ureteropelvic junction obstruction is present, pyeloplasty is the treatment of choice.
Fused pelvic kidney (pancake kidney)
Fused pelvic kidney is much less common. A single pelvic kidney is served by 2 collecting systems and ureters.
If obstruction is present, reconstruction is needed.
Kidney Malrotation
Malrotation of the kidney is usually of little clinical significance.
Ultrasound often shows hydronephrosis. Further evaluation with a magnetic resonance urogram or renal scan may be done when clinicians are concerned about possible obstruction.
Multicystic Dysplastic Kidney
In multicystic dysplastic kidney (MCDK), there is a nonfunctioning renal unit consisting of noncommunicating cysts with intervening solid tissue composed of fibrosis, primitive tubules, and foci of cartilage. Usually, ureteral atresia is also present. The contralateral kidney is usually normal, but up to 10% of patients may have vesicoureteral reflux or ureteropelvic junction obstruction. Frequently, the kidney progressively involutes and eventually is no longer visible on ultrasound. Development of tumors, infection, and/or hypertension is rare.
Most experts recommend observation to monitor for involution. Nephrectomy may be considered for the presence of solid tissue, progressive enlargement, or rarely hypertension or a ruptured cyst that is causing pain.
Renal Agenesis
Bilateral renal agenesis as part of a syndrome of oligohydramnios, pulmonary hypoplasia, and extremity and facial anomalies (classic Potter syndrome) is fatal within minutes to hours because of the pulmonary hypoplasia and subsequent respiratory distress. Fetal demise is common.
Unilateral renal agenesis occurs in approximately 1/2000 to 3000 births (1, 2). The true incidence is difficult to determine because many cases result from complete involution in utero of a multicystic dysplastic kidney. It usually is accompanied by ureteral agenesis with absence of the ipsilateral trigone and ureteral orifice. However, the ipsilateral adrenal gland is unaffected. No treatment is necessary; compensatory hypertrophy of the solitary kidney maintains normal renal function.
The kidneys share a common embryologic origin with the vas deferens and uterus. Boys may have agenesis of the vas deferens, and girls may have uterine anomalies. Girls may have an obstructed hemiuterus. At time of puberty, evaluation for uterine anomalies with a pelvic ultrasound should be considered for girls with significant dysmenorrhea, pelvic pain, and/or a pelvic mass (due to collection of blood) (3).
Renal agenesis references
1. Laurichesse Delmas H, Kohler M, Doray B, et al. Congenital unilateral renal agenesis: Prevalence, prenatal diagnosis, associated anomalies. Data from two birth-defect registries. Birth Defects Res. 2017;109(15):1204-1211. doi:10.1002/bdr2.1065
2. The Fetal Medicine Foundation. Renal agenesis. Accessed August 7, 2024.
3. Tan YG, Laksmi NK, Yap TL, Sadhana N, Ong CCP. Preventing the O in OHVIRA (Obstructed Hemivagina Ipsilateral Renal Agenesis): Early Diagnosis and Management of Asymptomatic Herlyn-Werner-Wunderlich Syndrome. J Pediatr Surg. 2020;55(7):1377-1380. doi:10.1016/j.jpedsurg.2019.06.006
Renal Dysplasia
In renal dysplasia (a histologic diagnosis), the renal vasculature, tubules, collecting ducts, or drainage apparatus develops abnormally.
Diagnosis of renal dysplasia is by biopsy.
If dysplasia is segmental, treatment of renal dysplasia is often unnecessary. If dysplasia is extensive, renal dysfunction may necessitate nephrologic care, including renal replacement therapy.
Renal Ectopia
Renal ectopia (abnormal renal location) usually results when a kidney fails to ascend from its origin in the true pelvis; a rare exception occurs with a superiorly ascended (thoracic) kidney. Pelvic ectopia increases the incidence of ureteropelvic junction obstruction, vesicoureteral reflux, and multicystic renal dysplasia.
Obstruction and severe reflux may be corrected surgically when indicated (if causing hypertension, recurrent infections, or renal growth retardation).
Renal Hypoplasia
Hypoplasia usually occurs because inadequate ureteral bud branching causes an underdeveloped, small kidney with histologically normal nephrons.
If hypoplasia is segmental, hypertension can occur, and ablative surgery may be needed. Patients should be evaluated for vesicoureteral reflux.





