



Von Hippel-Lindau disease is a rare hereditary neurocutaneous disorder characterized by benign and malignant tumors in multiple organs. Diagnosis is made using clinical criteria and/or molecular genetic testing. Treatment is with surgery or sometimes radiation therapy or, for retinal angiomas, laser coagulation or cryotherapy. Belzutifan and pazopanib are targeted therapies that reduce tumor size in patients with VHL-associated cancer.
Von Hippel-Lindau (VHL) disease is a neurocutaneous syndrome that is inherited as an autosomal dominant trait with variable penetrance.
The VHL gene is a tumor suppressor gene located on the short arm of chromosome 3 (3p25.3). Over 1500 different variants in this gene have been identified in patients with VHL disease. Both familial (inherited) and de novo cases occur (1). Mosaic forms have also been reported (2).
Variants in the VHL gene result in loss of translated VHL protein function and constitutive activation of hypoxia-inducible factor (HIF) pathways, which drive the development of highly vascular tumors in multiple organs.
In one Danish longitudinal cohort study, the pooled birth incidence of VHL disease was 1 in 27,300 people (3). A study of VHL disease from the United Kingdom estimated the birth incidence to be 1 in 36,000 people (4).
VHL disease most commonly causes:
Cerebellar hemangioblastomas
Retinal angiomas
Tumors, including pheochromocytomas and cysts (renal, hepatic, pancreatic, or genital tract), can occur in other organs. Approximately 4% of people with VHL disease develop an endolymphatic tumor in the inner ear, threatening hearing (5); however, this risk may be as high as 16% (1). The risk of developing renal cell carcinoma increases with age; the lifetime risk may be as high as 70% (6).
Manifestations typically appear between ages 10 and 30 but can appear earlier.
General references
1. van Leeuwaarde RS, Ahmad S, van Nesselrooij B, Zandee W, Giles RH. Von Hippel-Lindau Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; May 17, 2000.
2. Mikhaylenko DS, Kuryakova NB, Efremova AV, et al. Mosaic Form of von Hippel-Lindau Syndrome: Case Report and Literature Review. Int J Mol Sci. 2025;26(6):2751. Published 2025 Mar 19. doi:10.3390/ijms26062751
3. Binderup ML, Galanakis M, Budtz-Jørgensen E, Kosteljanetz M, Luise Bisgaard M. Prevalence, birth incidence, and penetrance of von Hippel-Lindau disease (vHL) in Denmark. Eur J Hum Genet. 2017;25(3):301-307. doi:10.1038/ejhg.2016.173
4. Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28(7):443-447. doi:10.1136/jmg.28.7.443
5. Bausch B, Wellner U, Peyre M, et al. Characterization of endolymphatic sac tumors and von Hippel-Lindau disease in the International Endolymphatic Sac Tumor Registry. Head Neck. 2016;38 Suppl 1:E673-E679. doi:10.1002/hed.24067
6. Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19(6):617-623. doi:10.1038/ejhg.2010.175
Symptoms and Signs of VHL Disease
The symptoms of VHL disease depend on the size and location of the tumors. Symptoms may include headaches, dizziness, hearing loss, weakness, ataxia, impaired vision, hematuria, flank pain, and hypertension.
Retinal angiomas appear as a dilated artery leading from the disk to a peripheral tumor with an engorged vein. These angiomas are usually asymptomatic, but if they are centrally located and enlarge, they can result in substantial loss of vision. These tumors increase the risk of retinal detachment, macular edema, and glaucoma.
Untreated, VHL disease can result in blindness, brain damage, or death. Death usually results from complications of cerebellar hemangioblastomas or renal cell carcinoma.
Diagnosis of VHL Disease
Direct ophthalmoscopy
Central nervous system imaging, typically MRI
Sometimes molecular genetic testing
Sometimes blood or urine testing
Sometimes audiometry
For the diagnosis of cancers associated with VHL disease, systematic screening of all at-risk organ systems should be performed starting in early childhood and continuing throughout life.
Several criteria for diagnosis (Dutch, Danish, and international consensus criteria) are available (1–3). VHL disease is diagnosed when 1 of the following criteria is met according to the Dutch or Danish criteria:
Family history of VHL disease and presence of ≥ 1 VHL tumor (retinal, brain, or spinal hemangioblastoma; pheochromocytoma; renal cell carcinoma; or pancreatic neuroendocrine tumor)
Two or more characteristic VHL tumors in patients with no known family history of VHL disease

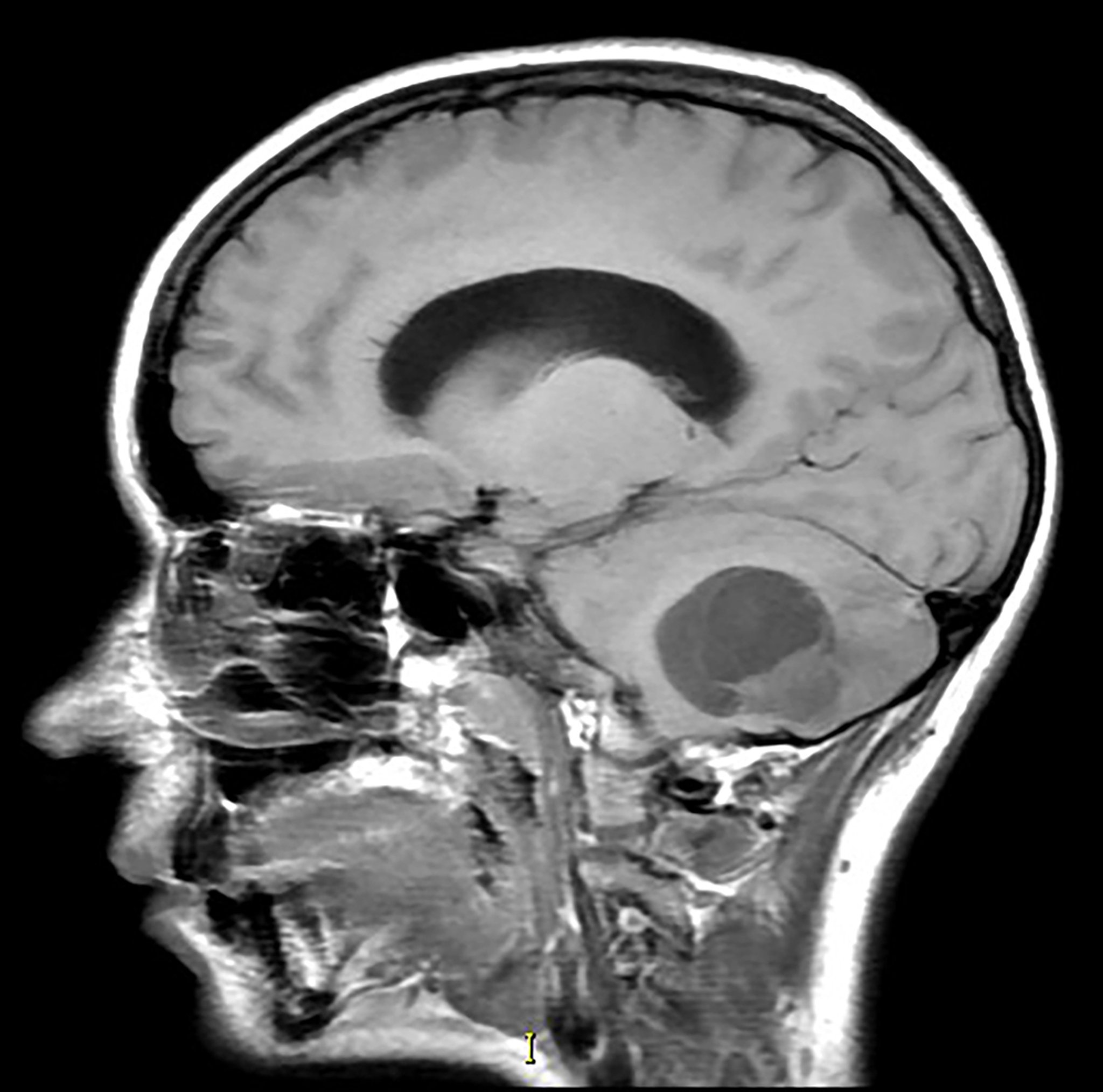
This sagittal T1-weighted magnetic resonance image without contrast shows a partially cystic and solid mass in the inferior cerebellum with associated mass effect. This is the most common appearance of a hemangioblastoma. This tumor is seen with increased frequency in patients with von Hippel-Lindau syndrome.
Living Art Enterprises/SCIENCE PHOTO LIBRARY
The diagnosis can be established by using molecular genetic testing for the presence of a heterozygous variant (pathogenic or likely pathogenic) in the VHL gene, even if clinical features are inconclusive.
Annual blood or urine tests for fractionated metanephrines are used for surveillance of pheochromocytomas.
Testing for hearing loss (particularly in patients who may have endolymphatic sac tumors) involves a complete audiometric evaluation.
If a specific variant for the VHL gene is identified in a patient, genetic testing should be done to determine whether at-risk family members also have that variant.
Diagnosis references
1. Louise M Binderup M, Smerdel M, Borgwadt L, et al. von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur J Med Genet. 2022;65(8):104538. doi:10.1016/j.ejmg.2022.104538
2. Hes FJ, van der Luijt RB, Lips CJ. Clinical management of Von Hippel-Lindau (VHL) disease. Neth J Med. 2001;59(5):225-234. doi:10.1016/s0300-2977(01)00165-6
3. Daniels AB, Tirosh A, Huntoon K, et al. Guidelines for surveillance of patients with von Hippel-Lindau disease: Consensus statement of the International VHL Surveillance Guidelines Consortium and VHL Alliance. Cancer. 2023;129(19):2927-2940. doi:10.1002/cncr.34896
Treatment of VHL Disease
Surgery or sometimes radiation therapy
Belzutifan in selected patients with renal cell carcinomas, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors
Pazopanib in patients with advanced renal cell carcinoma
For retinal angiomas, laser coagulation or cryotherapy
Regular monitoring
The treatment of VHL disease relies on tailored management strategies targeting specific tumor types. Treatment often involves surgical removal of the tumor before it becomes harmful. Pheochromocytomas are surgically removed; sometimes continued treatment of hypertension is needed. Renal cell carcinomas are surgically removed; advanced cancers may respond to pharmacologic treatment. Some tumors can be treated with focused high-dose radiation.
Belzutifan, an oral hypoxia-inducible factor-2 alpha inhibitor, is recommended for use in adult patients with VHL renal cell carcinomas (1), central nervous system hemangioblastomas (2), or pancreatic or neuroendocrine tumors (eg, pheochromocytomas) (3) that do not require immediate surgical removal (4). Belzutifan can be used until the disease progresses or unacceptable toxicity (eg, life-threatening hypoxia or anemia, extreme transaminitis) occurs.
Pazopanib, a multi-tyrosine kinase inhibitor (eg, receptors of vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor), is an alternative agent that has been shown to achieve a > 50% objective response rate in renal lesions in patients with VHL-associated advanced renal cell carcinoma (5).
Typically, retinal angiomas are treated with laser coagulation or cryotherapy to preserve vision.
Use of propranolol to reduce the size of the hemangiomas is being studied (6).
Screening to check for complications and instituting early treatment can improve prognosis.
Screening for complications
If the diagnostic criteria for VHL disease are met, patients should be regularly screened to check for complications of VHL disease because early detection is key to preventing serious complications.
Routine surveillance should include the following (7):
Annual history and physical examination
Annual dilated eye examination beginning at the age of diagnosis to screen for retinal hemangioblastomas
Annual blood pressure monitoring beginning at the age of diagnosis to screen for pheochromocytomas
Annual measurement of urine or plasma fractionated metanephrines beginning at 5 years of age to screen for pheochromocytomas
Brain and spinal MRI every 2 years beginning at 11 years of age to screen for central nervous system hemangioblastomas
Audiography every 2 years beginning at 11 years of age to screen for endolymphatic sac tumors
Abdominal MRI or ultrasound once after 15 years of age to screen for renal cell carcinomas, pheochromocytomas, and pancreatic neuroendocrine tumors
People who do not meet diagnostic criteria for VHL disease but who have a germline mutation or who have not been tested but are first- and second-degree family members of a patient with VHL disease also should be screened using the following:
Annual evaluation for neurologic symptoms and for vision and hearing problems
Annual ocular examination to look for nystagmus, strabismus, and white pupils
Annual blood pressure monitoring
Treatment references
1. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Neuroendocrine and Adrenal Tumors, version 3.2025. https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf. Accessed November 5, 2025.
2. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Central Nervous System Cancers, version 2.2025. https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf. Accessed November 5, 2025.
3. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Kidney Cancer, version 1.2026. https://www.nccn.org/professionals/physician_gls/pdf/neuroendocrine.pdf. Accessed November 5, 2025.
4. Srinivasan R, Iliopoulos O, Beckermann KE, et al. Belzutifan for von Hippel-Lindau disease-associated renal cell carcinoma and other neoplasms (LITESPARK-004): 50 months follow-up from a single-arm, phase 2 study. Lancet Oncol. 2025;26(5):571-582. doi:10.1016/S1470-2045(25)00099-3
5. Jonasch E, McCutcheon IE, Gombos DS, et al. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018;19(10):1351-1359. doi:10.1016/S1470-2045(18)30487-X
6. Albiñana V, Villar Gómez de Las Heras K, Serrano-Heras G, et al. Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients. Orphanet J Rare Dis. 2015;10:118. Published 2015 Sep 22. doi:10.1186/s13023-015-0343-5
7. Rednam SP, Becktell KD, Villani A, et al. Update on Surveillance in Von Hippel-Lindau Disease. Clin Cancer Res. 2025;31(12):2271-2277. doi:10.1158/1078-0432.CCR-24-3525
Key Points
Von Hippel-Lindau (VHL) disease is a rare hereditary neurocutaneous disorder characterized by benign and malignant tumors in multiple organs.
Most commonly, VHL disease causes cerebellar hemangioblastomas and retinal angiomas.
Diagnosis is confirmed if the patient has a family history of VHL disease and presence of ≥ 1 VHL tumor or has ≥ 2 characteristic VHL tumors with no known family history of VHL disease.
Molecular genetic testing can establish difficult to diagnose cases.
Treat with surgical removal of tumors and laser coagulation or cryotherapy for retinal angiomas.
Consider pazopanib for patients with advanced renal cell carcinoma and belzutifan for selected patients with renal cell carcinomas, central nervous system hemangioblastomas, or pancreatic or neuroendocrine tumors.
Regularly screen for complications, which can guide early treatment and improve prognosis.
Drug Information for the Topic





