



Idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia, causes progressive pulmonary fibrosis. Symptoms and signs develop over months to years and include exertional dyspnea, cough, and fine (Velcro) crackles. Diagnosis is based on history, physical examination, high-resolution CT, and/or lung biopsy, if necessary. Treatment may include antifibrotic drugs and oxygen therapy. Most patients deteriorate rapidly; median survival is about 3 to 5 years from diagnosis.
Idiopathic pulmonary fibrosis, identified histologically as usual interstitial pneumonia, accounts for most cases of idiopathic interstitial pneumonia. Prevalence varies widely, in part due to regions with more rigorous screening, with some estimates as high as 495 per 100,000 annually in the United States (1). IPF affects men and women > 50 years in a ratio of 2:1, with a markedly increased incidence with each decade of age (2). Current or former cigarette smoking is most strongly associated with the disorder. There is some genetic predisposition (especially MUC5B gene polymorphisms, which causes mucin overexpression); this may occur in approximately 30-35% of cases (3).
General references
1. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11 [published correction appears in Lancet Respir Med 2014 Jul;2(7):e12]. Lancet Respir Med 2014;2(7):566-572. doi:10.1016/S2213-2600(14)70101-8
2. Maher TM, Bendstrup E, Dron L, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res 2021;22(1):197. doi:10.1186/s12931-021-01791-z
3. Evans CM, Fingerlin TE, Schwarz MI, et al. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev 2016;96(4):1567-1591. doi:10.1152/physrev.00004.2016
Etiology and pathogenesis of Idiopathic Pulmonary Fibrosis
IPF is characterized by recurrent small injury to the alveolar epithelial cells, which are already predisposed to senescence (1). A combination of environmental (eg, cigarette smoking), genetic (eg, MUC5B gene polymorphisms), and other unknown factors probably contribute to alveolar epithelial cell dysfunction or reprogramming, which leads to abnormal fibroproliferation in the lung. Alterations in the lung microbiome leading to replication of certain species such as Prevotella, Veillonella, and Escherichia, may also contribute to pathology by affecting mucociliary clearance. Occupational exposures such as inhalation of toxic fumes can cause chemical damage and antibiotic use can alter the lung microbiome. Ultimately, a combination of these factors causes a shift in immune responses, leading to disease.
Etiology reference
1. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med 2018;378(19):1811-1823. doi:10.1056/NEJMra1705751
Pathology of Idiopathic Pulmonary Fibrosis
The characteristic histologic pattern of idiopathic pulmonary fibrosis is referred to as usual interstitial pneumonia (see table ). The key histologic findings include subpleural fibrosis with sites of fibroblast proliferation (fibroblast foci) and dense scarring, alternating with areas of normal lung parenchymal tissue (heterogeneity) (1). Scattered interstitial inflammation occurs with lymphocyte, plasma cell, and histiocyte infiltration. Cystic abnormality (honeycombing) occurs in all patients and increases with advanced disease. A similar histologic pattern uncommonly occurs in cases of interstitial lung diseases of known etiology.
Pathology reference
1. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
Symptoms and Signs of Idiopathic Pulmonary Fibrosis
Symptoms and signs of idiopathic pulmonary fibrosis typically develop over months to several years and include progressively worsening dyspnea on exertion and nonproductive cough, present in virtually every patient with IPF. Constitutional symptoms, such as low-grade fever and myalgias, are uncommon. The classic sign of IPF is fine, dry, inspiratory crackles (Velcro crackles) at both lung bases. Clubbing is present in about 50% of cases (1). Acrocyanosis is possible. The remainder of the examination is normal until disease is advanced, at which time signs of pulmonary hypertension and right ventricular systolic dysfunction may develop.
Symptoms and signs reference
1. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med 2018;378(19):1811-1823. doi:10.1056/NEJMra1705751
Diagnosis of Idiopathic Pulmonary Fibrosis
High-resolution CT (HRCT)
Sometimes surgical lung biopsy
Pulmonary function testing
Diagnosis of idiopathic pulmonary fibrosis is suspected in patients with subacute dyspnea, nonproductive cough, and Velcro crackles on chest examination. However, IPF is commonly overlooked initially because of clinical similarities to other more common diseases, such as bronchitis, asthma, and heart failure.
Diagnosis requires HRCT.
Chest radiograph typically shows diffuse reticular opacities in the lower and peripheral lung zones. Small cystic lesions (honeycombing) and dilated airways due to traction bronchiectasis are additional findings, usually occurring in more advanced cases with fibrosis.
HRCT shows diffuse, patchy, subpleural, reticular opacities with irregularly thickened interlobular septa and intralobular lines; subpleural honeycombing; and traction bronchiectasis. Significant ground-glass opacities, nodules, or mosaic attenuation/air trapping of the lung suggests an alternative diagnosis.
Pulmonary function testing usually reveals typical features of restrictive lung disease due to fibrosis. These are decreased diffusing capacity of the lung for carbon dioxide (DLCO), exertional hypoxia that progresses to low saturations at rest, and normal to low forced vital capacity (FVC). Reduced DLCO is seen in nearly every patient with IPF and helps confirm the diagnosis if other clinical and radiographic features of IPF are also present.
Lung biopsy may be considered if diagnostic uncertainty remains after the initial evaluation.

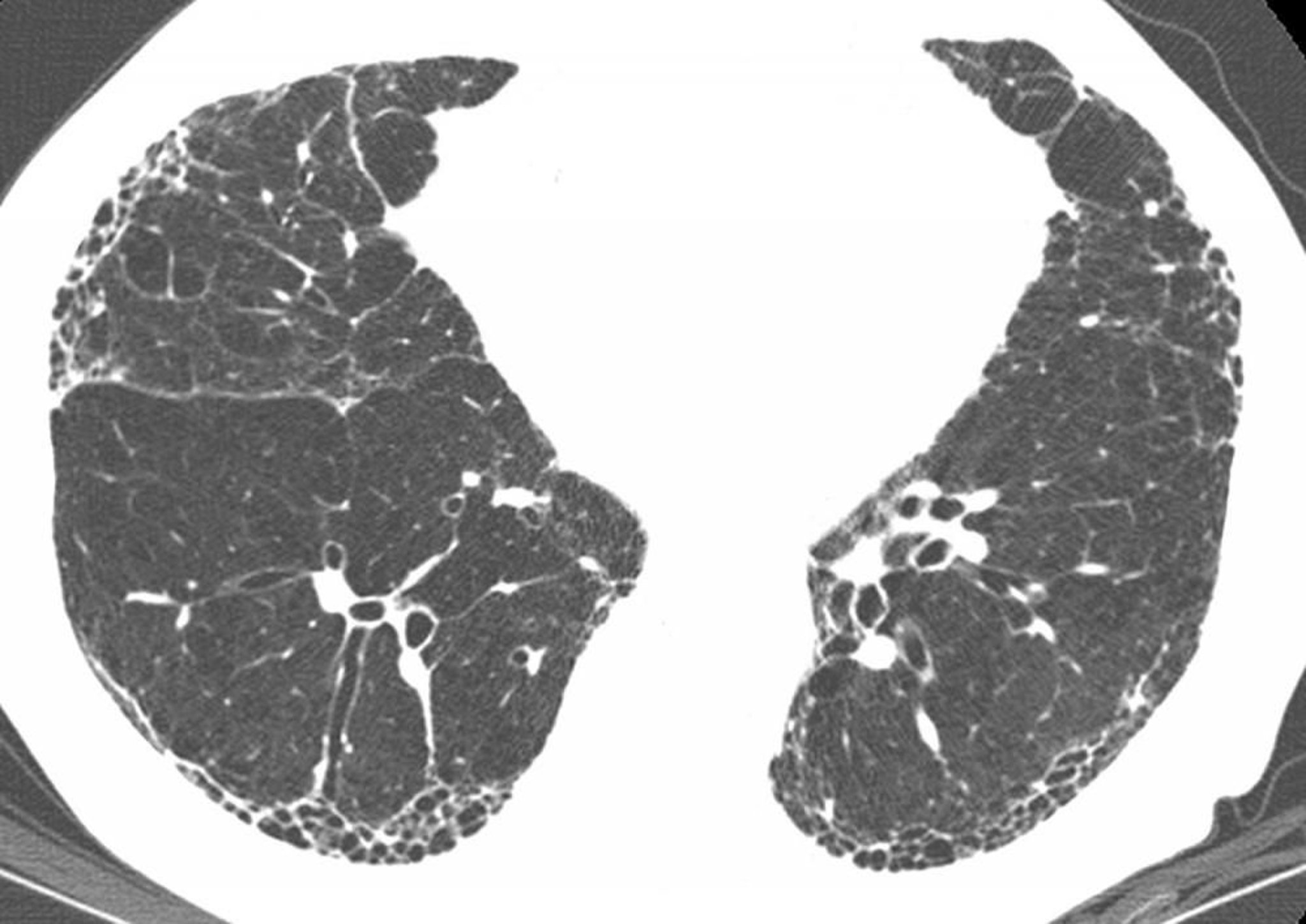
Subpleural honeycombing and traction bronchiectasis (signet ring sign, where multiple thickened dilated bronchi are seen below a pulmonary artery) are present on high-resolution CT in a patient with idiopathic pulmonary fibrosis (IPF). Findings inconsistent with IPF, such as nodules, are absent. These CT findings are virtually diagnostic of IPF if clinical findings are compatible.
Image courtesy of Harold R. Collard, MD.
Laboratory testing plays little role in diagnosis, except to exclude potential systemic rheumatic disorders.
Treatment of Idiopathic Pulmonary Fibrosis
Pirfenidone or nintedanib
Supplementary oxygen and pulmonary rehabilitation
Sometimes lung transplantation
Pirfenidone and nintedanib are antifibrotic medications that slow progression of idiopathic pulmonary fibrosis (1, 2, 3, 4). There is insufficient evidence to support the efficacy of one agent over the other. The use of these agents requires periodic monitoring of liver function tests.
Supportive measures include oxygen and pulmonary rehabilitation. Participating in a support group may help reduce the psychological stress associated with the illness.
Many novel therapies for IPF are under development or being tested as treatments for IPF, and patients should be encouraged to participate in clinical trials when appropriate.
Lung transplantation is successful for otherwise healthy patients with IPF. These patients should be evaluated for lung transplantation at the time of diagnosis.
Treatment references
1. Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis [published correction appears in Lancet Respir Med 2017 Jan;5(1):e7. doi: 10.1016/S2213-2600(16)30422-2.]. Lancet Respir Med 2017;5(1):33-41. doi:10.1016/S2213-2600(16)30326-5
2. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2022;205(9):e18-e47. doi:10.1164/rccm.202202-0399ST
3. Raghu G, Rochwerg B, Zhang Y, et al: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 192 (2):e3-e19, Jun 15, 2015.
4. Richeldi L, du Bois RM, Raghu G, et al: Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082, 2014.
Prognosis for Idiopathic Pulmonary Fibrosis
Most patients have moderate to advanced clinical disease at the time of diagnosis and deteriorate despite treatment. Antifibrotic therapy has decreased the mortality rate. Several prognostic models have been proposed. Among the factors that portend a worse prognosis are older age, male sex, lower forced vital capacity, and lower DLCO (1).
Causes of acute deterioration include infections, deep venous thrombosis, pulmonary embolism, pneumothorax, and heart failure. Pulmonary hypertension may also occur. Also, acute exacerbations without an identifiable cause may occur. Acute exacerbations tend to have a high morbidity and mortality, especially among patients who are hospitalized.
Lung cancer occurs more frequently in patients with IPF, but cause of death is usually respiratory failure. Because of the poor prognosis of IPF, early discussions with the patient and family about advance care planning and end-of-life care are important.
Prognosis reference
1. Sun X, Lei S, Zhao H, Guo L, Wang Y. Mortality-related risk factors of idiopathic pulmonary fibrosis: a systematic review and meta-analysis. J Thorac Dis 2024;16(12):8338-8349. doi:10.21037/jtd-23-1908
Key Points
Idiopathic pulmonary fibrosis accounts for most idiopathic interstitial pneumonia and tends to affect older people.
Symptoms and signs (eg, subacute dyspnea, nonproductive cough, and Velcro crackles) are nonspecific and usually caused by other, more common disorders.
High-resolution CT can help in diagnosis by showing findings such as diffuse, patchy, subpleural, reticular opacities with irregularly thickened interlobular septa and intralobular lines; subpleural honeycombing; and traction bronchiectasis.
Treat supportively and, if available, use pirfenidone or nintedanib.
Encourage participation in clinical trials and, if patients are otherwise healthy, consider lung transplant referral at the time of diagnosis.
Drug Information for the Topic





