



Sarcoidosis is an inflammatory disorder resulting in noncaseating granulomas in one or more organs and tissues; etiology is unknown. The lungs and lymphatic system are most often affected, but sarcoidosis may affect any organ. Pulmonary symptoms range from none to cough, exertional dyspnea and, rarely, lung or other organ failure. Diagnosis usually is first suspected because of pulmonary involvement on chest imaging (radiograph or CT scan) and is confirmed by biopsy and exclusion of other causes of granulomatous inflammation. The clinical course of sarcoidosis is heterogenous and unpredictable. The majority of the patients are asymptomatic at the time of diagnosis and go into remission without treatment. Treatment usually is indicated in symptomatic patients. First-line treatment is corticosteroids. Prognosis is excellent for limited disease but poor for more advanced disease.
Epidemiology of Sarcoidosis
Sarcoidosis most commonly affects people aged 20 to 40 years but occasionally affects children and older adults. Sarcoidosis has worldwide distribution, but prevalence is greatest in Black American people and ethnic northern European people, especially Scandinavian people. Disease presentation varies widely by racial and ethnic background, with Black American patients having more severe and frequent extrathoracic manifestations (1). Sarcoidosis is slightly more prevalent in women.
Epidemiology reference
1. Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis Based on Health Care Use. Ann Am Thorac Soc 2016;13(8):1244-1252. doi:10.1513/AnnalsATS.201511-760OC
Etiology of Sarcoidosis
Sarcoidosis is thought to be due to an exaggerated inflammatory response to certain antigen triggers in a genetically susceptible person (1, 2). Proposed triggers include
Infectious agents: Propionibacterium acnes and Mycobacteria (potentially the Mycobacterium tuberculosis catalase-peroxidase [mKatG] protein)
Environmental exposures: Mold or mildew and certain unidentified substances (organic and inorganic) present in workplaces with musty odors
Occupational exposures: Employment in metal (beryllium), agricultural and pesticide-using industries,ship workers, fire fighters and rescue workers who responded to the attacks on the World Trade Center (3)
Tobacco use is inversely correlated with sarcoidosis (2).
Evidence supporting genetic susceptibility includes the following (4):
Higher rate of disease concordance in monozygotic than dizygotic twins
Increased prevalence of sarcoidosis among first- or second-degree relatives of patients who have sarcoidosis
Marked increase in relative risk of developing sarcoidosis in siblings of patients who have sarcoidosis
Identification of several possible human leukocyte antigen (HLA) and non-HLA genes associated with sarcoidosis risk, course, and phenotypes
For example the HLA-DRB1*03/DQB1*02 haplotype is associated with Löfgren syndrome and predicts excellent prognosis, in contrast to HLA-DRB1*15/HLA DQB1*0602, which predicts persistent disease.
Etiology references
1. Chen ES, Moller DR. Etiologies of Sarcoidosis. Clin Rev Allergy Immunol 2015;49(1):6-18. doi:10.1007/s12016-015-8481-z
2. Newman LS, Rose CS, Bresnitz EA, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med 2004;170(12):1324-1330. doi:10.1164/rccm.200402-249OC
3. Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol 2012;12(2):145-150. doi:10.1097/ACI.0b013e3283515173
4. Iannuzzi MC. Advances in the genetics of sarcoidosis. Proc Am Thorac Soc 2007;4(5):457-460. doi:10.1513/pats.200606-136MS
Pathophysiology of Sarcoidosis
The unknown antigen triggers a cell-mediated immune response that is characterized by the accumulation of T cells and macrophages, release of cytokines and chemokines, including tumor necrosis factor alpha, and organization of responding cells into granulomas (1, 2). Clusters of disease in families and communities suggest a genetic predisposition, shared exposures, or, less likely, person-to-person transmission.
The inflammatory process leads to formation of noncaseating granulomas, the pathologic hallmark of sarcoidosis. Granulomas are collections of mononuclear cells and macrophages that differentiate into epithelioid and multinucleated giant cells and are surrounded by lymphocytes, plasma cells, fibroblasts, and collagen. Granulomas occur most commonly in the lungs and lymph nodes but can involve any organ and cause significant dysfunction. Granulomas in the lungs are distributed along lymphatics, with most occurring in peribronchiolar, subpleural, and perilobular regions. Granuloma accumulation distorts architecture in affected organs. Sarcoidal granulomas may persist, resolve, or lead to fibrosis. Whether granulomas lead directly to fibrosis or run a parallel course is not known.
Hypercalcemia may occur because of increased conversion of vitamin D to the activated form (1,25 hydroxy vitamin D) by macrophages. Hypercalciuria may be present, even in patients with normal serum calcium levels. Nephrolithiasis and nephrocalcinosis may occur, sometimes leading to chronic kidney disease.

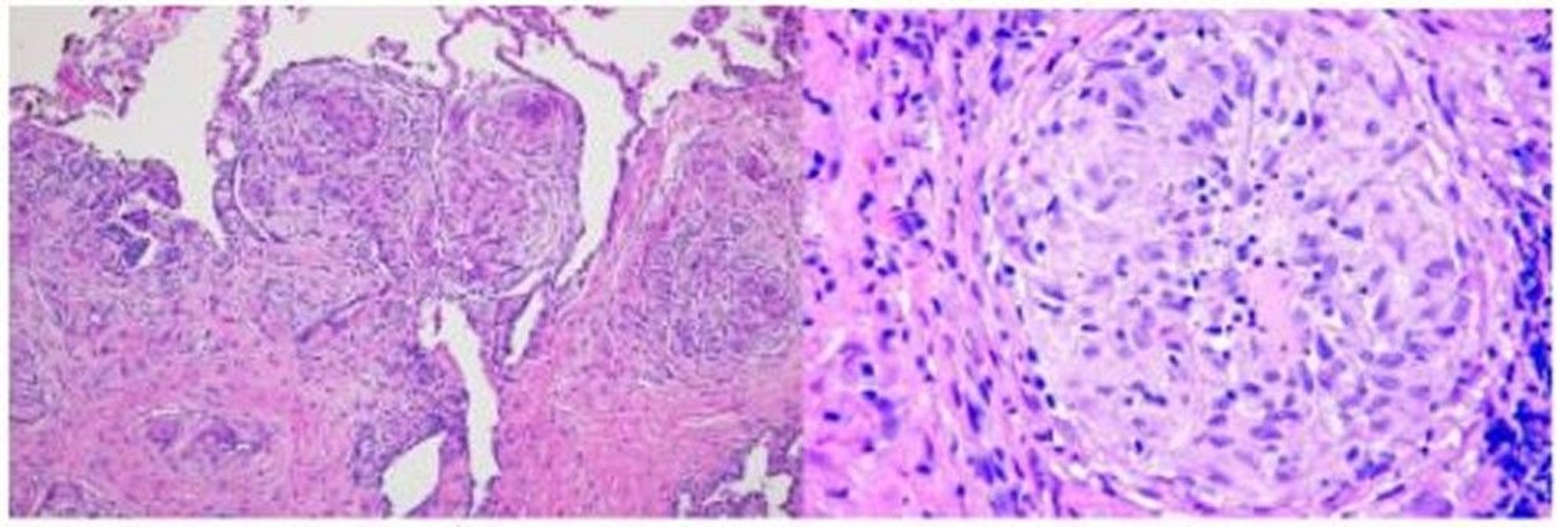
In this image, a lung biopsy of a patient with sarcoidosis shows granulomas along the bronchovascular bundle (hematoxylin and eosin stain, low magnification; left) and sarcoid granuloma with multinucleated giant cells in center of granulomas (high magnification; right).
Image courtesy of Birendra P. Sah, MD, FCCP.
Pathophysiology references
1. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011;183(5):573-581. doi:10.1164/rccm.201006-0865CI
2. Soler P, Basset F. Morphology and distribution of the cells of a sarcoid granuloma: ultrastructural study of serial sections. Ann N Y Acad Sci 1976;278:147-160. doi:10.1111/j.1749-6632.1976.tb47026.x
Symptoms and Signs of Sarcoidosis
Symptoms and signs depend on the site and degree of involvement and vary over time, ranging from spontaneous remission to chronic indolent illness. Accordingly, frequent reassessment for new symptoms and involvement in different organs is needed. Most cases are probably asymptomatic and thus go undetected. Pulmonary disease occurs in > 90% of adults with sarcoidosis (1, 2).
Symptoms and signs may include dyspnea, cough, chest discomfort, and crackles (3). Fatigue, malaise, weakness, anorexia, weight loss, and low-grade fever are also common. Sarcoidosis can manifest as fever of unknown origin.
Systemic involvement causes various symptoms (see table ), which vary by race, sex, and age. Black people are more likely than White people to have involvement of the eyes, liver, bone marrow, peripheral lymph nodes, and skin; erythema nodosum is an exception. Women are more likely to have erythema nodosum and eye or nervous system involvement. Men and older patients are more likely to have hypercalcemia.
Systemic Involvement in Sarcoidosis
System | Estimated Frequency | Comments |
|---|---|---|
Pulmonary | > 90% | Granulomas form in alveolar septa and bronchiolar and bronchial walls, causing diffuse pulmonary disease Rarely pulmonary arteries and veins can also be involved, causing pulmonary hypertension Often it is asymptomatic and detected incidentally on chest imaging (chest radiograph or CT scan) Spontaneously resolves in many patients but can cause progressive pulmonary dysfunction, leading to limitations in physical function, cavitary lesions, respiratory failure, and death in a few |
Constitutional | 80% | Fatigue, chronic pain, and fibromyalgia |
Dermatologic | 25% |
Common skin lesions:
Lupus pernio:
|
Lymph nodes
| Thoracic lymph nodes: 90% Extrathoracic lymph nodes: 15% | Hilar or mediastinal involvement in most patients Intra-abdominal or nontender to tender peripheral (neck, axilla or groin)l lymphadenopathy in few patients |
Hepatic | 12% | Usually asymptomatic Manifests with mild elevations in liver function test results, hypolucent lesions on CT scans with contrast Rarely, clinically significant cholestasis or cirrhosis Unclear distinction between sarcoidosis and granulomatous hepatitis when sarcoidosis affects the liver only |
Ocular | 12% | Uveitis (most common), causing blurred vision, photophobia, and tearing Can cause blindness Spontaneously resolves in most patients May manifest with conjunctivitis, iridocyclitis, chorioretinitis, dacryocystitis, lacrimal gland infiltration causing dry eyes, optic neuritis, glaucoma, or cataracts Ocular involvement more common among Black American patients and patients of Japanese descent Annual screening indicated for early disease detection |
Psychiatric | 10% | Depression (common), but uncertain whether it is a primary manifestation of sarcoidosis or a response to the prolonged course of disease and frequent recurrences |
Splenic | 10% | Usually asymptomatic Manifests with left upper quadrant pain and thrombocytopenia or as an incidental finding on ultrasound or CT |
Neurologic | < 10% | Cranial neuropathy, especially the 7th nerve (causing facial nerve palsy) or 8th nerve (causing hearing loss) Optic and peripheral neuropathy (common) May affect any cranial nerve Central nervous system involvement, with nodular lesions or diffuse meningeal inflammation typically in the cerebellum and brain stem Hypothalamic and pituitary stalk infiltration, possibly causing panhypopituitarism Arginine vasopressin deficiency, polyphagia and obesity, and thermoregulatory and libidinal changes |
Upper respiratory tract | < 10% | Acute and chronic granulomatous inflammation of nasal and sinus mucosa with symptoms indistinguishable from common allergic and infectious sinusitis; more common in patients with lupus pernio Laryngeal involvement present as nodule over vocal cords and hoarseness Tracheal involvement can lead to tracheal stenosis (rare) Diagnosis confirmed by biopsy |
Hematologic | < 5–30% | Anemia due to granulomatous infiltration of bone marrow, sometimes causing pancytopenia Splenic sequestration causing thrombocytopenia |
Bone | 5% | Osteolytic or cystic lesions, mostly asymptomatic Osteopenia |
Cardiac | < 5% | Conduction blocks and arrhythmias (most common), sometimes causing sudden death (cardiac sarcoidosis) Heart failure due to restrictive cardiomyopathy (primary) or Class V pulmonary hypertension (multifactorial) Transient papillary muscle dysfunction and pericarditis (rare) More common among patients of Japanese ancestry, in whom cardiomyopathy is the most frequent cause of sarcoidosis-related death |
Oral | < 5% | Asymptomatic parotid swelling (most common) Parotitis with xerostomia Heerfordt syndrome (uveoparotid fever), characterized by uveitis, bilateral parotid swelling, facial palsy, and chronic fever Oral lupus pernio, which may disfigure the hard palate and may involve the cheek, tongue, and gums |
Muscle | < 1% | Asymptomatic disease with or without enzyme elevations in most patients Sometimes insidious or acute myopathy with muscle weakness |
Joint | < 1% | Ankle, knee, wrist, and elbow arthritis (most common) May cause chronic arthritis with Jaccoud deformities or dactylitis Löfgren syndrome (triad of acute migratory polyarthritis, erythema nodosum, and hilar adenopathy) |
Renal | < 1% | Asymptomatic hypercalciuria (most common) Chronic kidney disease caused by nephrolithiasis and nephrocalcinosis due to chronic hypercalciuria and requiring renal replacement (dialysis or transplantation) in some patients |
Gastrointestinal tract | Rare | Most common gastric involvement by granulomas (asymptomatic or gastritis not responding to PPI) Oropharyngeal or esophageal dysphagia due to respective muscle involvement by granulomas Rarely, appendicular, colon, or rectal involvement Mesenteric lymphadenopathy that may cause abdominal pain |
Endocrine | Rare | Hypothalamic and pituitary stalk infiltration, possibly causing panhypopituitarism May cause thyroid infiltration without dysfunction Secondary hypoparathyroidism due to hypercalcemia |
Pleural | Rare | Causes lymphocytic exudative effusions, usually bilateral |
Reproductive | Rare | Case reports of endometrial, ovarian, epididymal, and testicular involvement No effect on fertility May subside during pregnancy and relapse postpartum |
CT = computed tomography; PPI = proton pump inhibitor | ||
Data from Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001;164(10 Pt 1):1885-1889. doi:10.1164/ajrccm.164.10.2104046; Carmona EM, Kalra S, Ryu JH. Pulmonary Sarcoidosis: Diagnosis and Treatment. Mayo Clin Proc 2016;91(7):946-954. doi:10.1016/j.mayocp.2016.03.004; and Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA 2011;305(4):391-399. doi:10.1001/jama.2011.10 | ||


This image shows dacryocystitis manifesting as inflammation of the palpebral lobe of the lacrimal glands in both upper eyelids in a patient with sarcoidosis.

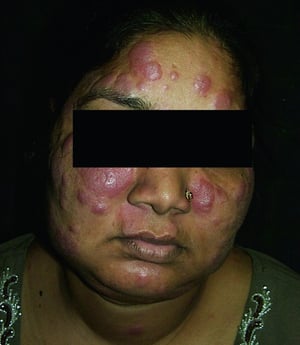
This photo shows erythematous facial plaques in a patient with sarcoidosis.
This photo shows erythematous facial plaques in a patient with sarcoidosis.
© Springer Science+Business Media

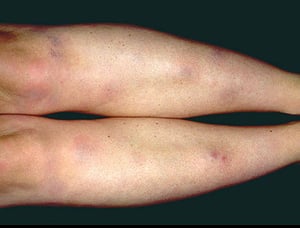
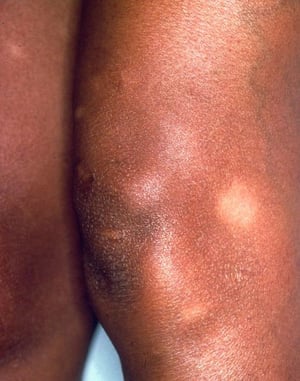
This photo shows characteristic tender, erythematous, subcutaneous nodules on the shins of a patient with erythema nodosum.
This photo shows characteristic tender, erythematous, subcutaneous nodules on the shins of a patient with erythema nodo
Photo provided by Thomas Habif, MD.

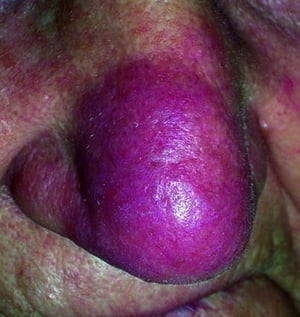
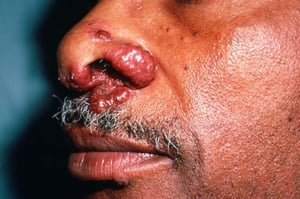
This photo shows violaceous plaques on the nose of a patient with sarcoidosis.
This photo shows violaceous plaques on the nose of a patient with sarcoidosis.
© Springer Science+Business Media

This image shows skin nodules in a patient with sarcoidosis.
This image shows skin nodules in a patient with sarcoidosis.
Image courtesy of Karen McKoy, MD.

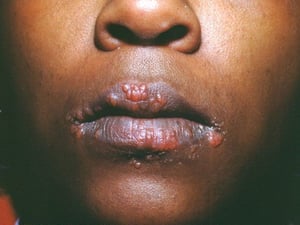
This image shows multiple papules on the lips of a patient with sarcoidosis.
This image shows multiple papules on the lips of a patient with sarcoidosis.
Image courtesy of Karen McKoy, MD.

Hypopigmented areas and subcutaneous nodules in a patient with sarcoidosis.
Hypopigmented areas and subcutaneous nodules in a patient with sarcoidosis.
Image courtesy of Karen McKoy, MD.
This photo shows erythematous facial plaques in a patient with sarcoidosis.
This photo shows erythematous facial plaques in a patient with sarcoidosis.
© Springer Science+Business Media
This photo shows characteristic tender, erythematous, subcutaneous nodules on the shins of a patient with erythema nodosum.
This photo shows characteristic tender, erythematous, subcutaneous nodules on the shins of a patient with erythema nodo
Photo provided by Thomas Habif, MD.
This photo shows violaceous plaques on the nose of a patient with sarcoidosis.
This photo shows violaceous plaques on the nose of a patient with sarcoidosis.
© Springer Science+Business Media
This image shows skin nodules in a patient with sarcoidosis.
This image shows skin nodules in a patient with sarcoidosis.
Image courtesy of Karen McKoy, MD.
This image shows multiple papules on the lips of a patient with sarcoidosis.
This image shows multiple papules on the lips of a patient with sarcoidosis.
Image courtesy of Karen McKoy, MD.
Hypopigmented areas and subcutaneous nodules in a patient with sarcoidosis.
Hypopigmented areas and subcutaneous nodules in a patient with sarcoidosis.
Image courtesy of Karen McKoy, MD.
Löfgren syndrome
Löfgren syndrome manifests as a triad of acute migratory polyarthritis, erythema nodosum, and hilar adenopathy (3). Fever, malaise, uveitis, and parotitis may also be present. Löfgren syndrome is more common among people of European ancestry. Löfgren syndrome is self-limited. Patients can usually be treated with nonsteroidal anti-inflammatory drugs (NSAIDs) alone. Rate of relapse is low.
Heerfordt syndrome
Heerfordt syndrome (uveoparotid fever) manifests as swelling of the parotid gland (due to sarcoid infiltration), uveitis, chronic fever, and less often palsy of the facial nerve. Heerfordt syndrome can be self-limited. Treatment is the same as for sarcoidosis.
Blau syndrome
Blau syndrome is a sarcoidosis-like disease inherited in an autosomal dominant fashion that manifests in children. It is not known whether Blau syndrome arises through the same mechanism as sarcoidosis diagnosed in adults. In Blau syndrome, children present before the age of 4 years with arthritis, rash, and uveitis. Blau syndrome is often self-limited. Symptoms usually are relieved with NSAIDs.
Symptoms and signs references
1. Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001;164(10 Pt 1):1885-1889. doi:10.1164/ajrccm.164.10.2104046
2. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA 2011;305(4):391-399. doi:10.1001/jama.2011.10
3. Carmona EM, Kalra S, Ryu JH. Pulmonary Sarcoidosis: Diagnosis and Treatment. Mayo Clin Proc 2016;91(7):946-954. doi:10.1016/j.mayocp.2016.03.004
Diagnosis of Sarcoidosis
Chest imaging
Biopsy
Exclusion of other granulomatous disorders
Sarcoidosis is most often suspected when hilar adenopathy, with or without lung infiltrates, is incidentally detected on chest radiography. Bilateral hilar adenopathy is the most common abnormality.
If sarcoidosis is suspected, a chest radiograph should be the first test if it has not already been done (1). The chest radiograph appearance tends to roughly predict the likelihood of spontaneous remission of pulmonary sarcoidosis (see table ). However, staging sarcoidosis by chest radiography can be misleading in regard to predicting disease severity; for example, extrapulmonary sarcoidosis, such as cardiac sarcoidosis or neurologic sarcoidosis, can portend a poor prognosis in the absence of evidence of pulmonary involvement. Also, chest radiograph findings do not correlate well with pulmonary function testing, and thus may not accurately indicate the severity of pulmonary sarcoidosis.
Chest Radiograph Staging of Sarcoidosis
Stage | Definition | Incidence of Spontaneous Remission |
|---|---|---|
0 | Normal chest radiograph | — |
I | Bilateral hilar lymphadenopathy without pulmonary infiltrates | 60–80% |
II | Bilateral hilar lymphadenopathy with pulmonary infiltrates | 50–65% |
III | Pulmonary infiltrates without bilateral hilar lymphadenopathy | < 30% |
IV | Pulmonary fibrosis | 0% |
Data from Consensus conference: activity of sarcoidosis. Third WASOG meeting, Los Angeles, USA, September 8-11, 1993. Eur Respir J 1994;7(3):624-627 | ||
A normal chest radiograph (stage 0) does not exclude the diagnosis of sarcoidosis, particularly when cardiac or neurologic involvement is suspected. A high-resolution CT is more sensitive for detecting hilar and mediastinal lymphadenopathy and parenchymal abnormalities. Lung parenchymal involvement is predominantly in the upper lobes but can be in any part of lungs. CT findings (see also image ) in more advanced radiographic stages (II to IV) include the following:
Thickening of the bronchovascular bundles and bronchial walls
Beading of the interlobular septa
Ground-glass opacification
Parenchymal nodules, cysts, or cavities
Traction bronchiectasis

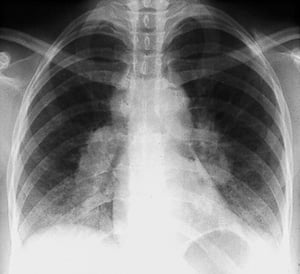
Bilateral hilar adenopathy in stage I sarcoidosis.
Bilateral hilar adenopathy in stage I sarcoidosis.
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

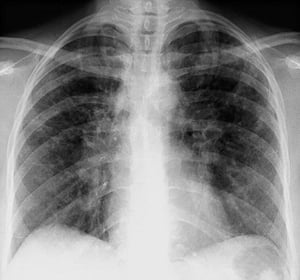
Bilateral hilar adenopathy with interstitial opacities in stage II sarcoidosis.
Bilateral hilar adenopathy with interstitial opacities in stage II sarcoidosis.
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

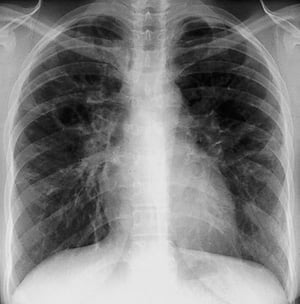
Diffuse interstitial opacities without hilar adenopathy in stage III sarcoidosis.
Diffuse interstitial opacities without hilar adenopathy in stage III sarcoidosis.
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

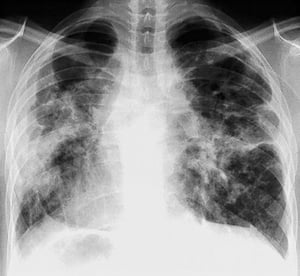
Severe, diffuse fibrosis with hilar adenopathy and cystic changes of the upper lobes in stage IV sarcoidosis.
Severe, diffuse fibrosis with hilar adenopathy and cystic changes of the upper lobes in stage IV sarcoidosis.
By permission of the publisher. From: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

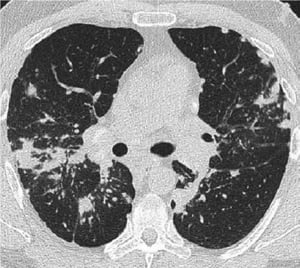
This high-resolution CT of the chest of a patient with pulmonary sarcoidosis shows thickening of the bronchovascular bundles and beading of the interlobular septa.
This high-resolution CT of the chest of a patient with pulmonary sarcoidosis shows thickening of the bronchovascular bu
Image courtesy of Birendra P. Sah, MD, FCCP.
Bilateral hilar adenopathy in stage I sarcoidosis.
Bilateral hilar adenopathy in stage I sarcoidosis.
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
Bilateral hilar adenopathy with interstitial opacities in stage II sarcoidosis.
Bilateral hilar adenopathy with interstitial opacities in stage II sarcoidosis.
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
Diffuse interstitial opacities without hilar adenopathy in stage III sarcoidosis.
Diffuse interstitial opacities without hilar adenopathy in stage III sarcoidosis.
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
Severe, diffuse fibrosis with hilar adenopathy and cystic changes of the upper lobes in stage IV sarcoidosis.
Severe, diffuse fibrosis with hilar adenopathy and cystic changes of the upper lobes in stage IV sarcoidosis.
By permission of the publisher. From: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
This high-resolution CT of the chest of a patient with pulmonary sarcoidosis shows thickening of the bronchovascular bundles and beading of the interlobular septa.
This high-resolution CT of the chest of a patient with pulmonary sarcoidosis shows thickening of the bronchovascular bu
Image courtesy of Birendra P. Sah, MD, FCCP.
When imaging suggests sarcoidosis, the diagnosis is confirmed by demonstration of noncaseating granulomas on biopsy and exclusion of alternative causes of granulomatous disease (see table ). As long as the patient does not have risk factors that suggest tuberculosis, Löfgren syndrome usually does not require confirmation by biopsy.
The diagnostic evaluation, therefore, requires the following (1):
Selection of a biopsy site
Exclusion of other causes of granulomatous disease
Assessment of the severity and extent of disease to determine whether therapy is indicated
Differential Diagnosis of Sarcoidosis
Type | Specific Disorder |
|---|---|
Mycobacterial infection | Atypical mycobacteria |
Fungal infection | |
Other infections | Cat-scratch disease (lymph nodes only) |
Rheumatologic disorders | Granulomatosis with polyangiitis Juvenile idiopathic arthritis (juvenile rheumatoid arthritis) Kikuchi-Fujimoto disease (lymph nodes only) |
Hematologic cancer | Castleman disease (a lymphoproliferative disorder associated with infection by HIV or human herpesvirus 8) |
Hypersensitivity | Metals encountered in occupational settings: Aluminum (appliances and equipment manufacture, construction industries), silica (construction, mining, foundries), beryllium (aerospace industries, nuclear reactors), titanium (medical implants, cosmetics, paints industries), zirconium (surgical appliances, glass, amalgam industries) Organic antigens causing hypersensitivity pneumonitis: Actinomycetes, atypical mycobacterial antigens, fungi, mushroom spores, other bioaerosols Inorganic antigens causing hypersensitivity pneumonitis: Isocyanate, pyrethrins Medication reaction (eg, minocycline, bleomycin, nitrofurantoin) Vaping induced |
Other | Foreign body aspiration or inoculation Granulomatous hepatitis |
Sites for biopsy
Appropriate biopsy sites may be obvious from physical examination and initial assessment; peripheral lymph nodes, skin lesions, and conjunctivae are all easily accessible. Endobronchial ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) of a mediastinal or hilar lymph node has a reported diagnostic yield of about 90% and is the diagnostic procedure of choice in patients with intrathoracic involvement (2).
When pulmonary sarcoidosis is suspected and EBUS-TBNA is nondiagnostic, bronchoscopic transbronchial lung biopsy with bronchoalveolar lavage (BAL) can be used. It can also be used in patients without any lung parenchymal infiltrates because diagnostic yield for transbronchial lung biopsy in stage I sarcoidosis is about 50% (3). If the bronchoscopic transbronchial biopsy is nondiagnostic, it can be tried a second time.
If EBUS-TBNA and bronchoscopic transbronchial biopsies are nondiagnostic or if bronchoscopy cannot be tolerated, mediastinoscopy can be done to biopsy mediastinal or hilar lymph nodes, or video-assisted thoracoscopic (VAT) lung biopsy or open-lung biopsy can be done to obtain lung tissue. If sarcoidosis is strongly suspected but a biopsy site is not evident based on examination or imaging findings, positron emission tomography (PET) scanning can help identify the occult active sites such as bone, muscle, liver, or spleen.
Exclusion of other diagnoses
Exclusion of other diagnoses is critical, especially when symptoms and radiographic signs are minimal, because many other disorders and processes can cause granulomatous inflammation (see table ). Biopsy tissue should be processed for fungi and mycobacteria tests. Exposure history to occupational (eg, silica, beryllium), environmental (eg, moldy hay, birds, and other antigenic triggers of hypersensitivity pneumonitis), and infectious (eg, tuberculosis, coccidioidomycosis, histoplasmosis) antigens should be explored. Purified protein derivative (PPD) skin testing or interferon gamma release assay should be done early in the assessment.
Disease severity assessment
Severity is assessed according to organ involvement so, for example, with only pulmonary involvement
Pulmonary function tests
Pulmonary function test results are often normal in early stages but demonstrate restriction and reduced diffusing capacity for carbon monoxide (DLCO) in advanced disease. Airflow obstruction also occurs and may suggest involvement of the bronchial mucosae. Adding a 6-minute walk test may characterize functional impairment more comprehensively than the results of pulmonary function tests alone. Patients with extensive lung involvement may have normal oxygen saturation at rest but may show desaturation with exertion.
Recommended routine screening tests for extrapulmonary disease include:
Baseline 12- lead ECG, echocardiography, and Holter monitoring only if symptoms of palpitation or syncope
Slit-lamp ophthalmologic examination
Routine blood tests to evaluate renal and hepatic function
Serum calcium levels and 24-hour urinary calcium excretion
Complete blood cell count with differential
Imaging
Imaging is often needed to detect extrapulmonary sarcoidosis.
Cardiac magnetic resonance imaging (MRI) with and without gadolinium contrast may be appropriate in patients with cardiac symptoms.
In patients with neurologic symptoms, brain or spine MRI with or without gadolinium may be needed.
Electromyography may be appropriate in patients with symptoms of peripheral neuropathy.
PET scanning appears to be the most sensitive test for detecting bone and other extrapulmonary sarcoidosis and is used together with MRI in patients with cardiac involvement.
Abdominal CT with radiopaque contrast agents is not routinely recommended but can provide evidence of hepatic or splenic involvement (eg, enlargement, hypolucent lesions).
Whole-body gallium scanning has been largely replaced by PET scanning. If available, gallium scanning may provide useful supportive evidence in the absence of tissue confirmation. Symmetric increased uptake in mediastinal and hilar nodes (lambda sign) and in lacrimal, parotid, and salivary glands (panda sign) strongly suggest sarcoidosis. A negative result in patients taking prednisone is unreliable.
Laboratory testing
Laboratory testing plays an adjunctive role in establishing the diagnosis and determining the extent of organ involvement.
Complete blood count with differential may show anemia, eosinophilia, leukopenia, or thrombocytopenia. Serum calcium should be measured to detect hypercalcemia.
Blood urea nitrogen (BUN), creatinine, and liver test results may be elevated in renal and hepatic sarcoidosis. Total protein may be elevated because of hypergammaglobulinemia.
Elevated erythrocyte sedimentation rate and C-reactive protein are common but nonspecific. In a patient with history of kidney stones, measurement of calcium in a urine specimen collected over 24 hours is recommended to exclude hypercalciuria, even if serum calcium level is normal.
Elevated serum angiotensin-converting enzyme (ACE) levels can suggest sarcoidosis but are nonspecific and may be elevated in patients with other conditions (eg, hyperthyroidism, diabetes, Gaucher disease, silicosis, mycobacterial disease, fungal infections, hypersensitivity pneumonitis, lymphoma). Angiotensin-converting enzyme (ACE) levels, if elevated, may be useful for monitoring adherence with corticosteroid treatment. ACE levels plummet even when patients are taking low-dose corticosteroids.
Bronchoalveolar lavage
Bronchoalveolar lavage should be done along with bronchoscopic biopsy to rule out suspected infections (eg, when findings with less invasive means such as imaging are not typical of sarcoidosis) and to exclude other forms of interstitial lung disease if the diagnosis of sarcoidosis is in doubt. The findings on bronchoalveolar lavage vary considerably, but lymphocytosis (lymphocytes > 15%), a CD4+/CD8+ ratio of > 3.5 in the lavage fluid cell differential, or both suggest the diagnosis in the proper clinical context. However, absence of these findings does not exclude sarcoidosis.
Diagnosis references
1. Crouser ED, Maier LA, Wilson KC, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med 2020;201(8):e26-e51. doi:10.1164/rccm.202002-0251ST
2. Tremblay A, Stather DR, MacEachern P, Khalil M, Field SK. A randomized controlled trial of standard vs endobronchial ultrasonography-guided transbronchial needle aspiration in patients with suspected sarcoidosis. Chest 2009;136(2):340-346. doi:10.1378/chest.08-2768
3. Akten HS, Kilic H, Celik B, et al. Diagnostic Yield of Transbronchial Biopsy in Comparison to High Resolution Computerized Tomography in Sarcoidosis Cases. Asian Pac J Cancer Prev 2018;19(4):1029-1033. doi:10.22034/APJCP.2018.19.4.1029
Treatment of Sarcoidosis
Nonsteroidal anti-inflammatory drugs (NSAIDs)
Corticosteroids
Other immunosuppressants
Tumor necrosis factor antibodies
Because sarcoidosis often spontaneously resolves, asymptomatic patients and patients with mild symptoms do not require treatment, although they should be monitored for the signs of deterioration. These patients can be followed with serial chest radiographs, pulmonary function tests (including diffusing capacity), and markers of extrathoracic involvement (eg, routine renal and liver function testing, annual slit-lamp ophthalmologic examination). The frequency of follow-up testing is determined by the severity of disease.
Patients who require treatment regardless of chest radiograph stage include those with the following:
Worsening symptoms
Limitation of activity
Markedly abnormal or deteriorating lung functions
Worrisome radiographic changes (eg, cavitation, fibrosis, conglomerate masses, signs of pulmonary hypertension)
Heart, nervous system, or eye involvement
Renal or hepatic failure
Moderate to severe hypercalcemia
Disfiguring skin (eg, lupus pernio) or joint disease
Mild hypercalcemia can be treated with a low-calcium diet, hydration, and minimizing sunlight exposure.
NSAIDs are used to treat erythema nodosum and mild musculoskeletal discomfort.
Corticosteroids
Symptom management usually begins with corticosteroids (1). The presence of chest imaging abnormalities without significant symptoms or evidence of decline in organ function is not an indication for treatment.
A standard protocol is prednisone 20 mg to 40 mg by mouth once a day, depending on symptoms and severity of findings. Alternate-day regimens may be used, eg, prednisone 40 mg by mouth once every other day. Although patients rarely require > 40 mg/day, higher doses may be needed to reduce complications in severe neurologic and cardiac disease.
Response usually occurs within 6 to 12 weeks, so symptoms, other disease-severity markers, and pulmonary function test results may be reassessed between 6 and 12 weeks. Chronic, insidious cases may respond more slowly. Corticosteroids are tapered to a maintenance dose (eg, prednisone 10 to 15 mg/day) after evidence of response and are continued for an additional 6 to 9 months if improvement occurs.
The optimal duration of treatment is unknown. Premature taper can result in relapse. The medication is slowly stopped if response is absent or equivocal. Corticosteroids can ultimately be stopped in most patients, but because relapse occurs up to 50% of the time, monitoring should be repeated, usually every 3 to 6 months. Corticosteroid treatment should be resumed for recurrence of symptoms and signs. Because angiotensin-converting enzyme (ACE) production is suppressed with low doses of corticosteroids, serial serum ACE levels may be useful in assessing adherence with corticosteroid treatment in patients who have elevated ACE levels.
Inhaled corticosteroids can relieve cough in patients with endobronchial involvement. An inhaled bronchodilator can be added in patients with obstructive airway disease.
Topical corticosteroids may be useful in dermatologic, nasal sinus, and ocular disease.
Prophylaxis against Pneumocystis jirovecii pneumonia is recommended while patients are taking > 20 mg prednisone daily or its equivalent for more than a month and for those who are taking immunosuppressants.
Alendronate or another bisphosphonate may be the treatment of choice for prevention of corticosteroid-induced osteoporosis in people at risk (eg, older patients). Using supplemental calcium or vitamin D risks hypercalcemia due to endogenous production of active vitamin D (1, 25 dihydroxy vitamin D) by sarcoidal granulomas. Serum and 24-hour urinary calcium measurements should be normal before starting such supplements.
Pearls & Pitfalls
|
Other immunosuppressants
Other immunosuppressants are used when
Patients cannot tolerate prednisone
Sarcoidosis is refractory to moderate to high doses of prednisone
Prednisone dose cannot be tapered below 10 to 15 mg daily after 3 months
Prior to adding other immunosuppressants, possible reasons for lack of clinical improvement, such as nonadherence, comorbid disease (eg, asthma, heart failure, anemia), pulmonary hypertension, and end-stage fibrosis should be considered.
Methotrexate is the most commonly used immunosuppressant. Patients should be given a 6-month trial of methotrexate 10 to 15 mg/week. Before starting methotrexate, patients should be tested for hepatitis B virus and hepatitis C virus infection.
Initially, methotrexate and corticosteroids are both given; over 6 to 8 weeks, the corticosteroid dose can be tapered and, in many cases, stopped. The maximal response to methotrexate, however, may take 6 to 12 months. In such cases, prednisone must be tapered more slowly. Serial blood counts and liver enzyme tests should be done every 2 to 4 weeks initially and then every 6 to 12 weeks once a stable dose is achieved. Folic acid (1 mg by mouth once a day) is recommended for patients treated with methotrexate to reduce the risk of adverse effects.
Other nonbiologic immunosuppressants include azathioprine, mycophenolate, cyclophosphamide, leflunomide, and hydroxychloroquine. Hydroxychloroquine is usually effective for treating hypercalcemia, arthralgia, skin sarcoidosis, or enlarged uncomfortable or disfiguring peripheral lymph nodes. Ophthalmologic evaluation should be done before hydroxychloroquine is started and then every 12 months during treatment to monitor for its ocular toxicity.
Relapse is common after stopping an immunosuppressant.
Tumor necrosis factor inhibitors
Infliximab is usually used to treat refractory sarcoidosis and to treat patients who are intolerant to both corticosteroid and the above-mentioned nonbiologic immunosuppressants. Before beginning therapy, patients should have a purified protein derivative (PPD) or interferon gamma release assay to screen for latent tuberculosis. Infliximab is given intravenously and it may take up to 3 to 6 months for maximal response. Infliximab is usually combined with low-dose methotrexate or azathioprine to prevent antibody formation against it.
Adalimumab can be considered for patients with ocular or cutaneus sarcoidosis and in patients who have been treated successfully with infliximab but have developed antibodies or infusion reactions..
Other treatment considerations
Patients who have heart block or ventricular arrhythmias due to cardiac involvement should receive an implantable cardiac defibrillator and pacemaker in addition to pharmacotherapy.
Tetracycline antibiotics such as doxycycline or minocycline can be tried for cutaneous sarcoidosis (2).
No available medications have consistently prevented pulmonary fibrosis.
Treatment of sarcoidosis-associated pulmonary arterial hypertension (SPAH) is supportive with diuresis and supplemental oxygen. The role of pulmonary vasodilators in treating SPAH has not been well established; a few small studies have suggested efficacy, but larger studies are needed to confirm it (3).
Organ transplantation is an option for patients with end-stage pulmonary, cardiac, or liver involvement, although disease may recur in the transplanted organ.
Patients with sarcoidosis who have moderate or severe impairment of pulmonary or cardiac function and who are treated with immunosuppressants are at increased risk of developing severe viral infections (eg, influenza, respiratory syncytial virus [RSV], COVID-19) as well as pneumococcal pneumonia. Vaccines against these infections are strongly recommended in patients with sarcoidosis because these vaccines could reduce the mortality and severity of illness.
Treatment references
1. Baughman RP, Valeyre D, Korsten P, et al: ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J 58(6):2004079, 2021. doi:10.1183/13993003.04079-2020
2. Bachelez H, Senet P, Cadranel J, Kaoukhov A, Dubertret L: The use of tetracyclines for the treatment of sarcoidosis. Arch Dermatol 137(1):69-73, 2001. doi:10.1001/archderm.137.1.69
3. Humbert M, Kovacs G, Hoeper MM, et al: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61(1): 1-144, 2023. doi:10.1183/13993003.00879-2022
Prognosis for Sarcoidosis
Although spontaneous remission is common, disease manifestations and severity are highly variable, and many patients require corticosteroids to relieve symptoms or slow progressive organ function decline at some time during the disease course. Thus, serial monitoring for evidence of relapse is imperative. Almost two-thirds of patients with sarcoidosis eventually achieve remission with few or no sequelae (1). In about 50% of patients who have spontaneous remission, remission occurs within the first 3 years after diagnosis. Fewer than 10% of these patients relapse after 2 years. Patients who do not experience remission within 2 to 3 years are likely to have chronic disease.
Sarcoidosis is thought to be chronic in up to 30% of patients, and 10 to 20% experience permanent sequelae (1). The disease is fatal in less than 10% of patients, typically due to respiratory failure caused by pulmonary complications followed by severe cardiac involvement (2). Infiltrative cardiomyopathy causing arrhythmias and heart failure is a common cause of death.
Prognosis is worse for patients with extrapulmonary sarcoidosis and for Black patients.
Good prognostic signs include
Löfgren syndrome (triad of acute polyarthritis, erythema nodosum, and hilar adenopathy)
Poor prognostic signs include
Chronic uveitis
Lupus pernio
Chronic hypercalcemia
Neurosarcoidosis
Cardiac involvement
Extensive pulmonary involvement and/or development of pulmonary hypertension
Little difference is demonstrable in long-term outcome between treated and untreated patients, and relapse is common when treatment ends.
Prognosis references
1. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357(21):2153-2165. doi:10.1056/NEJMra071714
2. Drent M, Crouser ED, Grunewald J. Challenges of Sarcoidosis and Its Management. N Engl J Med 2021;385(11):1018-1032. doi:10.1056/NEJMra2101555
Key Points
Systemic and extrapulmonary involvement is common with sarcoidosis, but > 90% of adult patients have pulmonary involvement.
Obtain a chest imaging study but confirm the diagnosis by biopsy, usually endobronchial ultrasound-guided transbronchial needle aspiration of a mediastinal or hilar lymph node.
Assess pulmonary severity with pulmonary function testing and pulse oximetry during exercise.
Test for extrapulmonary involvement with ECG, slit-lamp examination, renal and hepatic function tests, and serum and urinary calcium testing.
Treat patients with systemic corticosteroids when indicated (eg, severe symptoms, hypercalcemia, progressive decline in organ function, cardiac or neurologic involvement).
Treat with other immunosuppressants if patients cannot tolerate moderate doses of corticosteroids, sarcoidosis is resistant to corticosteroids, or if corticosteroids are required long term.
Drug Information for the Topic


