




El mieloma múltiple es un cáncer de células plasmáticas que producen una inmunoglobulina monoclonal e invaden y destruyen el tejido óseo adyacente. Las manifestaciones frecuentes incluyen lesiones líticas en los huesos que causan dolor y/o fracturas, insuficiencia renal, hipercalcemia, anemia e infecciones recurrentes. El diagnóstico exige generalmente demostrar proteína M (que a veces está presente en orina y no en suero, pero rara vez está totalmente ausente) y/o proteinuria de cadenas ligeras y exceso de células plasmáticas en médula ósea. El tratamiento específico más a menudo incluye una combinación de la quimioterapia convencional, corticosteroides, y uno o más medicamentos adicionales como inhibidores del proteasoma (p. ej., bortezomib, carfilzomib, ixazomib), inmunomoduladores (p. ej., lenalidomida, talidomida, pomalidomida) o anticuerpos monoclonales (p. ej., daratumumab, isatuximab, elotuzumab). Los enfoques basados en anticuerpos y células T paraactuar contra el antígeno de maduración de células B han demostrado ser eficaces. También se puede utilizar melfalán en dosis altas seguido de alotrasplante de células madre de sangre periférica.
(Véase también Generalidades sobre los trastornos de las células plasmáticas).
En los Estados Unidos, el riesgo de desarrollar mieloma múltiple durante toda la vida es de 1 en 132 (0,76%) (1). La mediana de la edad es de 70 años. La prevalencia en individuos de raza negra es del doble que la observada en los de raza blanca. Hay un ligero predominio masculino. Se desconoce la etiología, aunque se han sugerido factores cromosómicos y genéticos, radiación y sustancias químicas.
La American Cancer Society estima que en 2023 en los Estados Unidos se diagnosticarán alrededor de 35.730 casos nuevos de mieloma múltiple y se esperan alrededor de 12.590 muertes (1).
Referencia general
1. American Cancer Society. Key Statistics About Multiple Myeloma. Atlanta, Ga., American Cancer Society; 2023.
Fisiopatología del mieloma múltiple
La proteína M (proteína inmunoglobulina monoclonal) producida por las células plasmáticas malignas es IgG en alrededor del 55% de los pacientes con mieloma e IgA en alrededor del 20%. De los pacientes que producen IgG o IgA, el 40% también tiene proteinuria de Bence Jones, que es la presencia de cadenas livianas kappa (κ) o lambda (λ) monoclonales, libres, en orina. En el 15-20% de los casos, las células plasmáticas secretan sólo proteína de Bence Jones. Los mielomas IgM e IgD representan alrededor del 1% de los casos cada uno; el mieloma IgD es más frecuente en pacientes de ascendencia asiática. El mieloma por IgE es extremadamente raro. En raras ocasiones, los pacientes no tienen proteína M en la sangre y en la orina, aunque el ensayo utilizado en la actualidad para medir la cadena ligera libre en suero demuestra cadenas ligeras monoclonales en muchos de estos pacientes antes considerados no secretores.
A menudo hay osteoporosis difusa o lesiones osteolíticas aisladas, con mayor frecuencia en la pelvis, la columna vertebral, las costillas, el fémur, el húmero y el cráneo. Las lesiones son causadas por el reemplazo óseo secundario a los plasmocitomas en expansión o a citocinas secretadas por las células plasmáticas malignas que activan a los osteoclastos e inhiben a los osteoblastos, lo que conduce a la pérdida ósea. Por lo general, las lesiones osteolíticas son múltiples; a veces son masas intramedulares solitarias. El aumento de la pérdida ósea también puede provocar hipercalcemia. Los plasmocitomas extraóseos solitarios son inusuales, pero pueden localizarse en cualquier tejido, en especial en las vías respiratorias superiores.
En muchos pacientes se presenta insuficiencia renal en el momento del diagnóstico o se desarrolla durante su evolución. La insuficiencia renal tiene numerosas causas y principalmente es el resultado del depósito de cadenas lgeras en los túbulos distales (enfermedad renal relacionada con el mieloma) o la hipercalcemia.
Asimismo, los pacientes a menudo desarrollan anemia debida a nefropatía o a la inhibición de la eritropoyesis por células cancerosas pero a veces también debido a otras causas no relacionadas, como deficiencia de hierro o deficiencia vitamina B12.
Debido a la falta de anticuerpos normales y otras disfunciones inmunitarias, algunos pacientes tienen mayor susceptibilidad a la infección bacteriana. Las infecciones virales, especialmente las infecciones por herpes zóster, aparecen como resultado de modalidades terapéuticas como inhibidores del proteasoma (p. ej., bortezomib, ixazomib, carfilzomib) y anticuerpos monoclonales (p. ej., daratumumab, elotuzumab, isatuximab).
La amiloidosis afecta al 10% de los pacientes con mieloma, la mayoría de las veces pacientes con proteínas M de tipo lambda.
Hay expresiones variantes de mieloma múltiple (véase tabla Expresiones variantes de mieloma múltiple).
Síntomas y signos del mieloma múltiple
El dolor óseo persistente (en especial en la espalda o el tórax), la insuficiencia renal y las infecciones bacterianas recurrentes son los problemas más frecuentes en el momento de la presentación, pero muchos casos se detectan cuando las pruebas de laboratorio de rutina muestran aumento de la concentración de proteínas totales en sangre, proteinuria o anemia o insuficiencia renal de causa desconocida.
Los síntomas de anemia predominan o pueden ser el único motivo de evaluación en algunos casos, y unos pocos pacientes presentan manifestaciones de síndrome de hiperviscosidad.
Las fracturas patológicas (fragilidad) (es decir, fracturas que ocurren con traumatismos mínimos o sin ningún traumatismo) son comunes, y el colapso vertebral puede provocar compresión de la médula espinal y paraplejía.
Son frecuentes la neuropatía periférica, el síndrome del túnel carpiano (en especial con enfermedad por amiloide asociada), el sangrado anormal y los síntomas de hipercalcemia (p. ej., polidipsia, deshidratación).
Las linfadenopatías y la hepatoesplenomegalia son infrecuentes.
Diagnóstico del mieloma múltiple
Hemograma completo con plaquetas, frotis de sangre periférica, y pruebas químicas (nitrógeno ureico en sangre, creatinina, calcio, ácido úrico, LDH)
Electroforesis de proteínas en suero y orina (en una muestra de orina de 24 horas), seguida de inmunofijación; inmunoglobulinas cuantitativas; cadenas livianas libres en suero
Radiografías (evaluación esquelética) y una tomografía por emisión de positrones (PET)-TC o una RM de cuerpo entero
Examen de médula ósea, que incluye estudios citogenéticos convencionales y de hibridación fluorescente in situ (FISH)
Se sospecha mieloma múltiple en pacientes > 40 años con dolor óseo sin causa reconocida, en particular durante la noche o en reposo, otros síntomas característicos o alteraciones de laboratorio inexplicables (como aumento de proteínas en sangre o en orina, hipercalcemia, insuficiencia renal o anemia) o radiografías que muestran una fractura patológica o lesiones líticas.
La evaluación de laboratorio incluye pruebas habituales en sangre, LDH, beta-2 microglobulina sérica, inmunoelectroforesis y electroforesis de proteínas en suero y orina, concentración sérica de cadenas ligeras libres. Los pacientes también deben someterse a una evaluación esquelética y, dado que son más sensible a la enfermedad ósea que las radiografías, a una PET-TC o una RM de cuerpo entero. También se requiere un examen de médula ósea junto con estudios citogenéticos convencionales y FISH (para obtener una revisión, véase [1, 2]).
Las pruebas en sangre de rutina son hemograma completo y determinaciones químicas. Se detecta anemia en el 80% de los pacientes, en general anemia normocítica-normocrómica con formación en "pilas de monedas" de eritrocitos, grupos de 3 a 12 eritrocitos. Los recuentos de leucocitos y plaquetas suelen ser normales. Puede haber un aumento del nitrógeno ureico en sangre, creatinina sérica, LDH, beta-2 microglobulina, y ácido úrico. En ocasiones, el hiato aniónico es bajo. Alrededor del 10% de los pacientes presentan hipercalcemia en el momento del diagnóstico.
La inmunoelectroforesis y la electroforesis de proteínas se realiza en una muestra de suero y en una muestra de orina concentrada de una recolección de 24 horas para cuantificar la concentración urinaria de proteína M. La electroforesis sérica identifica la proteína M en alrededor del 80 al 90% de los pacientes. La mayoría del 10 al 20% restante son pacientes con solo cadenas livianas monoclonales libres (proteína de Bence Jones) que pueden detectarse mediante electroforesis de proteínas en la orina e inmunitaria. En raras ocasiones, los pacientes tienen enfermedad por IgM, IgD o IgE. La proteína monoclonal no es evidente en el momento del diagnóstico en una pequeña proporción de pacientes; sin embargo, durante el curso de la enfermedad, más pacientes tienen evidencia de proteína monoclonal.
La electroforesis con inmunofijación puede identificar la clase de inmunoglobulina de la proteína M (IgG, IgA o pocas veces IgD, IgM o IgE) y, a menudo, puede detectar proteína de cadenas livianas si la inmunoelectroforesis en suero es falso-negativa; se realiza la electroforesis con inmunofijación aunque la prueba sérica sea negativa si hay una firme sospecha de mieloma múltiple.
El análisis de cadenas livianas libres en suero con identificación de las relaciones kappa y lambda o las diferencias entre las cadenas livianas involucradas y no comprometidas ayuda a confirmar el diagnóstico y también se puede utilizar para controlar la eficacia de la terapia y proporcionar datos pronósticos.
Si se confirma el diagnóstico o este es muy probable se mide la concentración de beta-2-microglobulina en suero y junto con la albúmina sérica se utiliza para estadificar a los pacientes como parte del sistema de estadificación internacional (véase tabla Sistema de estadificación internacional para el mieloma múltiple). La beta-2 microglobulina es una proteína pequeña que se encuentra sobre la membrana de todas las células. Su concentración varía directamente con la masa tumoral y la gravedad de la disfunción renal.
Los estudios radiológicos incluyen un estudio del esqueleto (es decir, radiografías simples de cráneo, huesos largos, columna vertebral, pelvis y costillas). En el 80% de los casos, se observan lesiones líticas en sacabocados u osteoporosis difusa. Por lo general, las gammagrafías óseas no son útiles. La RM de todo el cuerpo puede proporcionar más detalles y se obtiene si hay sitios específicos con dolor o síntomas neurológicos. La PET-TC proporciona información pronóstica y puede ayudar a determinar si los pacientes tienen plasmocitoma solitario o mieloma múltiple.

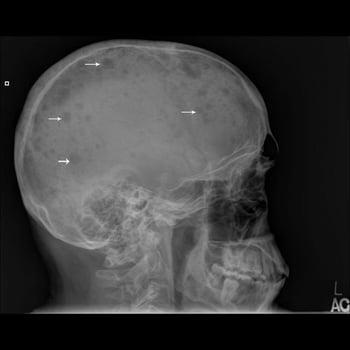
Se lleva a cabo una aspiración y una biopsia de la médula ósea, que muestran láminas o grupos de células plasmáticas; se diagnostica mieloma cuando ≥ 10% de las células son de este tipo. Sin embargo, el compromiso de la médula ósea es irregular; por lo tanto, algunas muestras de pacientes con mieloma pueden mostrar < 10% de células plasmáticas. Aun así, el porcentaje de células plasmáticas en la médula ósea rara vez es normal. La morfología de las células plasmáticas no se correlaciona con la clase de inmunoglobulina sintetizada. Los estudios cromosómicos de la médula ósea (p. ej., con técnicas de evaluación citogenéticas como hibridación in situ con fluorescencia [FISH] e inmunohistoquímica) pueden revelar anomalías cariotípicas específicas de las células plasmáticas que pueden influir sobre las opciones de tratamiento y que se asocian con diferencias en la supervivencia.
El diagnóstico y la diferenciación de otros tumores malignos (p. ej., carcinoma metastásico, linfoma, leucemia) y la gammapatía monoclonal de significado incierto normalmente requieren múltiples criterios:
Células plasmáticas clonales de la médula ósea o plasmacitoma
Proteína M en plasma, orina o ambos
Afectación orgánica (hipercalcemia, insuficiencia renal, anemia o lesiones óseas)
En pacientes con proteína M sérica, el mieloma está indicado por proteinuria de Bence Jones > 200 mg/24 horas o cadenas livianas libres anormales en suero, lesiones osteolíticas (sin evidencia de cáncer metastásico ni enfermedad granulomatosa) y láminas o grupos de células plasmáticas en la médula ósea.
Referencias del diagnóstico
1. Sive J, Cuthill K, Hunter H, Kazmi M, Pratt G, Smith D and on behalf of British Society of Haematology: Guidelines on the diagnosis, investigation and initial treatment of myeloma: a British Society for Haematology/UK Myeloma Forum Guideline. Brit J Haematol 193:245–268, 2021. doi:10.1111/bjh.17410
2. Rajkumar SV: Multiple myeloma: 2022 update on diagnosis, risk-stratification and management. Am J Hematol 97(8):1086-1107, 2022. doi:10.1002/ajh.26590
Pronóstico del mieloma múltiple
El mieloma múltiple es progresivo e incurable, pero la mediana de la supervivencia a 5 años ha mejorado hasta el 58% como resultado de los avances en el tratamiento (1). Los signos de pronóstico desfavorables en el momento del diagnóstico son un estadio más alto, niveles más bajos de albúmina sérica, niveles más altos de beta-2 microglobulina, niveles elevados de LDH, anomalías citogenéticas específicas en las células tumorales y niveles más altos de células malignas en la sangre. Los pacientes que presentan al inicio insuficiencia renal también tienen mala evolución, a menos que la función renal mejore con el tratamiento, lo que generalmente sucede con las opciones terapéuticas actuales.
Como el mieloma múltiple finalmente es fatal, a los pacientes se les debe ofrecer asesoramiento sobre cuidados terminales, con participación de médicos, familiares y amigos adecuados. Se deben incluir las instrucciones médicas por adelantado, el uso de sondas alimentarias y el alivio del dolor.
Referencia del pronóstico
1. American Cancer Society: Multiple Myeloma: Early Detection, Diagnosis, and Staging. Survival Rates for Multiple Myeloma. Atlanta, Ga., American Cancer Society; 2023.
Tratamiento del mieloma múltiple
Quimioterapia convencional para pacientes sintomáticos
Talidomida, lenalidomida o pomalidomida y/o bortezomib, carfilzomib o ixazomib con corticosteroides y/o quimioterapia convencional
Anticuerpos monoclonales, que incluyen elotuzumab, isatuximab y daratumumab
En particular para el mieloma recidivante o refractario, el inhibidor selectivo de la exportación nuclear (SINE) selinexor, y el inhibidor de la histona desacetilasa, panobinostato
En particular para el mieloma recidivante o refractario, tratamientos inmunológicos que se dirigen contra el antígeno de maduración de las células B (BCMA), muy expresado en las células de mieloma
El inhibidor de BCL-2 (regulador de la apoptosis de BCL2) venetoclax en pacientes cuyas células malignas presentan el marcador genético t(11;14)
Terapia de mantenimiento con corticosteroides, talidomida, y/o lenalidomida e inhibidores del proteasoma, en especial ixazomib por vía oral
Posiblemente alotrasplante de células madre
Posiblemente radioterapia de las áreas sintomáticas específicas que no responden a la terapia sistémica
Tratamiento de las complicaciones (anemia, hipercalcemia, insuficiencia renal, infecciones, y lesiones esqueléticas [en particular aquellas asociadas con alto riesgo de fractura])
El tratamiento del mieloma ha mejorado en las 2 últimas décadas, y la supervivencia a largo plazo es un objetivo terapéutico razonable. Consiste en el tratamiento directo de las células malignas en pacientes sintomáticos o aquellos con disfunción orgánica relacionada con el mieloma (anemia, disfunción renal, hipercalcemia o enfermedad ósea).
Los factores de riesgo para el requerimiento de tratamiento rápido del mieloma en pacientes que presentan en un principio disfunción de órganos incluyen
> 60% de células plasmáticas en la médula ósea
> 1 lesión en la RM
Concentraciones séricas de cadenas ligeras libres > 100 mg/L
Se considera que los pacientes con estos factores de riesgo tienen mieloma activo y requieren tratamiento inmediato, incluso aunque casi todos los ensayos clínicos aleatorizados sobre tratamiento temprano de estos pacientes no han demostrado un aumento de la supervivencia global. Es probable que los pacientes sin estos factores de riesgo ni disfunción de órganos terminales no se beneficien con la terapia inmediata, que suele diferirse hasta que aparecen síntomas o complicaciones.
Tratamiento de las células malignas
En el pasado, la quimioterapia convencional era el tratamiento inicial del mieloma múltiple, que consistía en melfalán por vía oral y prednisona administrados en 8 a 12 ciclos de 4 a 6 semanas con evaluación mensual de la respuesta. Sin embargo, se han logrado resultados superiores con la adición de un inhibidor del proteasoma, como bortezomib, carfilzomib o ixazomib, o los agentes inmunomoduladores lenalidomida o talidomida. Otros agentes antineoplásicos, incluidos ciclofosfamida, bendamustina, doxorrubicina y su análogo doxorrubicina liposómica pegilada (doxorrubicina liposómica), también son más eficaces cuando se los combina con un agente inmunomodulador (talidomida, lenalidomida o bortezomib). Se ogra mayor supervivencia cuando el tratamiento inicial incluye bortezomib y lenalidomida con corticosteroides. El agregado del anticuerpo monoclonal daratumumab al bortezomib y la dexametasona como parte del tratamiento inicial parece mejorar los resultados (1).
La respuesta al tratamiento (véase tabla Definición de la respuesta al tratamiento oncológico) se indica a través de
Disminución de las proteínas M séricas y urinarias
Disminución de los niveles séricos de la cadena ligera libre
Aumentos del número de glóbulos rojos
Mejora de la función renal en pacientes con insuficiencia renal
Normalización de los niveles de calcio entre los que presentan niveles elevados
Disminución del dolor óseo
Disminución de la fatiga
Puede considerarse el autotrasplante de células madre de sangre periférica en pacientes que tienen función cardíaca, hepática, pulmonar y renal adecuadas, en particular aquellos cuya enfermedad es estable o ha respondido después de varios ciclos de tratamiento inicial. Sin embargo, las nuevas opciones de tratamiento son muy eficaces y pueden hacer que el trasplante sea menos necesario o completamente innecesario. Ensayos clínicos recientes muestran una mayor supervivencia sin progresión, pero sin mejoría en la supervivencia global cuando los pacientes se someten a trasplante de células madre como parte de la terapia inicial (2).
El alotrasplante de células madre después de quimioterapia no mieloablativa (p. ej., ciclofosfamida y fludarabina en dosis bajas) o radioterapia en dosis baja puede determinar supervivencia de 5 a 10 años en algunos pacientes. Sin embargo, el trasplante alogénico de células madre con quimioterapia mieloablativa o no mieloablativa sigue siendo experimental debido a la alta morbimortalidad derivadas de la enfermedad de injerto contra huésped.
Tratamiento del mieloma recidivante o refractario
En el mieloma recidivante o resistente al tratamiento, puede recurrirse a combinaciones de un inhibidor del proteosoma (bortezomib, ixazomib o carfilzomib) con un inmunomodulador (talidomida, lenalidomida o pomalidomida) y quimioterapia o corticosteroides. Por lo general, estos fármacos se combinan con otros eficaces que el paciente todavía no ha recibido, aunque aquellos con remisiones prolongadas pueden responder a un nuevo tratamiento con el mismo esquema que indujo la remisión inicial. Los pacientes que no responden a una determinada combinación de drogas pueden responder cuando se sustituye por otro medicamento de la misma clase (p. ej., los inhibidores del proteasoma, agentes inmunomoduladores, fármacos quimioterapéuticos).
Los anticuerpos monoclonales dirigidos contra proteínas en las células de mieloma también pueden ser muy eficaces en el mieloma recidivante o refractario. Los anticuerpos monoclonales incluyen daratumumab, isatuximab y elotuzumab. Estos anticuerpos son más eficaces cuando se combinan con los agentes inmunomoduladores talidomida, con lenalidomida o pomalidomida y dexametasona. El daratumumab produce mejores resultados cuando se combina con bortezomib y dexametasona (3), y tanto el daratumumab como el isatuximab muestran una mayor eficacia cuando se agregan a carfilzomib y dexametasona. Sin embargo, el tratamiento con cualquiera de estos anticuerpos monoclonales y solo dexametasona es eficaz en muchos pacientes sin los costos y los efectos adversos de los medicamentos adicionales.
El inhibidor selectivo de la exportación nuclear (SINE) selinexor, y el inhibidor de la histona desacetilasa (HDACi), panobinostato son especialmente eficaces cuando se combinan con otros fármacos activos contra el mieloma.
Existen tres tratamientos inmunitarios eficaces dirigidos al antígeno de maduración de las células B (BCMA). Estos tratamientos incluyen terapias celulares para el mieloma, las terapias de las células T del receptor del antígeno quimérico (CAR) con idecabtagene vicleucel y ciltacabtagene autoleucel, y un anticuerpo biespecífico teclistamab que también se dirige contra CD3 en las células T. Aunque estos tratamientos son eficaces, pueden causar efectos adversos agudos significativos (síndrome de liberación de citocinas, problemas neurológicos) y un alto riesgo de infecciones graves persistentes.
Se ha demostrado que el inhibidor oral de BCL-2 venetoclax es eficaz para el tratamiento de pacientes cuyas células de mieloma muestran el marcador genético t(11;14) (4).
Terapia de mantenimiento
Se ha probado el tratamiento de mantenimiento con fármacos no antineoplásicos, como el interferón alfa, que prolonga la remisión pero no mejora la supervivencia y se asocia con efectos adversos importantes. Después de una respuesta a esquemas basados en corticoides, los corticoides solos son eficaces como tratamiento de mantenimiento.
La talidomida también puede ser eficaz como tratamiento de mantenimiento, y la lenalidomida sola o con corticosteroides es eficaz como tratamiento de mantenimiento. Sin embargo, surgió cierta preocupación en etapa por la malignidad secundaria entre los pacientes que reciben tratamiento con lenalidomida a largo plazo, sobre todo después del alotrasplante de células madre. Por lo tanto, el riesgo de desarrollar cánceres secundarios debe evaluarse en comparación con la mayor supervivencia.
El inhibidor del proteasoma por vía oral ixazomib es eficaz como agente único en la terapia de mantenimiento. Se desconoce si la combinación de ixazomib con lenalidomida es más eficaz.
El papel de los anticuerpos en la terapia de mantenimiento aún debe definirse.
Tratamiento de las complicaciones
Además del tratamiento directo de las células malignas, éste también debe dirigirse a las complicaciones, que incluyen
Anemia
Hipercalcemia
Hiperuricemia
Infecciones
Insuficiencia renal
Lesiones esqueléticas
La anemia puede tratarse con eritropoyetina recombinante en pacientes cuya anemia es aliviada inadecuadamente por la quimioterapia. Si la anemia causa síntomas cardiovasculares o sistémicos significativos, se transfunden concentrados de eritrocitos. La plasmaféresis está indicada si el paciente desarrolla hiperviscosidad, lo que rara vez ocurre en pacientes con mieloma. A menudo, los pacientes tienen deficiencia de hierro por razones no relacionadas con su mieloma y requieren hierro intravenoso. Los pacientes con anemia deben someterse a mediciones periódicas de los niveles séricos de hierro, transferrina y ferritina para controlar los depósitos de hierro, así como de vitamina B12.
La hipercalcemia se trata mediante saluresis intensiva, bisfosfonatos IV (preferiblemente ácido zoledrónico) después de rehidratación y, a veces con calcitonina o prednisona. El denosumab también puede usarse para tratar la hipercalcemia, especialmente entre los pacientes con insuficiencia renal grave. Los pacientes deben evitar los alimentos que contienen calcio, los suplementos de calcio y la vitamina D.
La hiperuricemia puede ocurrir en algunos pacientes con alta carga tumoral y problemas metabólicos subyacentes. Sin embargo, la mayoría de los pacientes no requieren alopurinol. El alopurinol o el rasburicasa se indica en pacientes con altas concentraciones séricas de ácido úrico o alta carga tumoral y alto riesgo de síndrome de lisis tumoral con el tratamiento.
Las infecciones son más probables durante la neutropenia inducida por quimioterapia. Además, las infecciones por el virus herpes zóster son frecuentes con algunos fármacos antimieloma, especialmente los inhibidores del proteosoma bortezomib, carfilzomib o ixazomib y los anticuerpos monoclonales (daratumumab, isatuximab, elotuzumab). Los tratamientos más nuevos dirigidos contra el antígeno de maduración de células B (BCMA) se asocian con un riesgo muy alto de infección grave.
Las infecciones bacterianas documentadas deben tratarse con antibióticos. El uso profiláctico de antibióticos no se recomienda de manera sistemática.
Puede estar indicada la administración profiláctica de fármacos antivirales (p. ej., aciclovir, valganciclovir, famciclovir) en pacientes que reciben un inhibidor del proteosoma (bortezomib, carfilzomib o ixazomib) o un anticuerpo monoclonal (daratumumab, isatuximab, elotuzumab).
La inmunoglobulina IV profiláctica puede reducir el riesgo de infección, pero en general se reserva para pacientes con bajos niveles de inmunoglobulinas inespecíficas e infecciones recurrentes frecuentes.
Se indican la vacuna antineumocócica y antigripal para prevenir la infección, pero estas no son eficaces en la mayoría de los pacientes debido a la inmunodeficiencia relacionada con la enfermedad y el tratamiento. No se recomienda el uso de vacunas a gérmenes vivos en pacientes inmunocomprometidos. A diferencia de la primera vacuna contra el zóster a virus vivos atenuados, la vacuna contra el zóster recombinante a virus no viables puede administrarse para prevenir el herpes zóster, pero también tiene una eficacia limitada.
El compromiso renal a menudo puede mejorar con hidratación adecuada. Aun pacientes con proteinuria de Bence Jones masiva y prolongada (≥ 10-30 g/día) pueden tener función renal intacta si mantienen una diuresis > 2.000 mL/día. La deshidratación combinada con agente de contraste IV hiperosmolar puede precipitar insuficiencia renal oligúrica en pacientes con proteinuria de Bence Jones. La plasmaféresis puede ser eficaz en algunos casos. Deben evitarse los fármacos nefrotóxicos. El tratamiento rápido y agresivo del mieloma subyacente para reducir los niveles de la inmunoglobulina monoclonal nefrotóxica es importante para revertir esta afección.
Las lesiones esqueléticas requieren múltiples medidas de sostén. El mantenimiento de la deambulación y los suplementos de calcio y vitamina D ayudan a preservar la densidad ósea. Los niveles de vitamina D deben ser medidos al momento del diagnóstico y de forma periódica y la dosificación de la vitamina D debe ajustarse en consecuencia. Los analgésicos y las dosis paliativas de radioterapia (18-24 Gy) pueden aliviar el dolor óseo. Sin embargo, la radioterapia puede provocar una toxicidad importante y dado que suprime la función medular, puede deteriorar la capacidad del paciente de recibir dosis citotóxicas de quimioterapia sistémica.
La mayoría de los pacientes, en especial aquellos con lesiones líticas y osteoporosis u osteopenia generalizada, deben recibir un bisfosfonato IV en forma mensual (pamidronato o ácido zoledrónico). Los bisfosfonatos reducen las complicaciones esqueléticas y el dolor óseo, y pueden ejercer un efecto antitumoral. Para los pacientes con insuficiencia renal potencialmente reversible secundaria a mieloma pero no relacionada con hipercalcemia o con reacciones inminentes a la infusión de bisfosfonatos, una opción es el denosumab mensual (administrado por vía subcutánea), que, a diferencia de los bifosfonatos, no es eliminado por el riñón y no causa reacciones a la infusión. Tanto los bisfosfonatos como el denosumab pueden causar osteonecrosis de la mandíbula con escasa frecuencia. Mantener una excelente salud dental y evitar los explantes y los implantes dentales son importantes para minimizar el riesgo de esta complicación.
Referencias del tratamiento
1. Abdallah N, Kumar SK. Daratumumab in untreated newly diagnosed multiple myeloma. Ther Adv Hematol 2019;10:2040620719894871. doi:10.1177/2040620719894871
2. Richardson PG, Jacobus SJ, Weller EA, et al. Triplet Therapy, Transplantation, and Maintenance until Progression in Myeloma. N Engl J Med 2022;387(2):132-147. doi:10.1056/NEJMoa2204925
3. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med 2016;375(8):754-766. doi:10.1056/NEJMoa1606038
4. Sidiqi MH, Al Saleh AS, Kumar SK, et al. Venetoclax for the treatment of multiple myeloma: Outcomes outside of clinical trials. Am J Hematol 2021;96(9):1131-1136. doi:10.1002/ajh.26269
Conceptos clave
Las células plasmáticas malignas producen una inmunoglobulina monoclonal e invaden y destruyen el hueso.
Los plasmocitomas en expansión y la secreción de citocinas causa múltiple lesiones osteolíticas separadas (por lo general en la pelvis, columna vertebral, costillas y cráneo) y osteoporosis difusa; son frecuentes el dolor, las fracturas y la hipercalcemia.
Es frecuente la anemia y la insuficiencia renal.
La amiloidosis se desarrolla en aproximadamente el 10%, por lo general en los pacientes que producen exceso de cadenas ligeras lambda.
Efectuar una electroforesis de proteínas en suero y orina, seguida de inmunofijación, medición cuantitativa de inmunoglobulinas y de cadenas livianas libres en suero.
Realizar aspiración y biopsia de médula ósea.
Los pacientes sintomáticos y aquellos con disfunción orgánica deben ser tratados con terapia farmacológica, que puede incluir corticosteroides, fármacos quimioterápicos, inhibidores del proteasoma, agentes inmunomoduladores, anticuerpos monoclonales, inhibidores selectivos de la exportación nuclear, inhibidores de la histona desacetilasa e inmunoterapia celular y humoral contra el antígeno de maduración de las células B.
El trasplante de células madre es una opción para algunos pacientes, pero las opciones terapéuticas más nuevas y eficaces pueden evitarlo en otros pacientes.
Más información
Los siguientes recursos en inglés pueden ser útiles. Tenga en cuenta que el MANUAL no es responsable por el contenido de estos recursos.
Bal S, Giri S, Godby KN, Costa LJ: New regimens and directions in the management of newly diagnosed multiple myeloma. Am J Hematol 96:367–378, 2021. doi:10.1002/ajh.26080
Cowan AJ, Green DJ, Kwok M, et al: Diagnosis and management of multiple myeloma: A review. JAMA 327:464-477, 2022. doi: 10.1001/jama.2022.0003
Cook G, Morris CTCM: Evolution or revolution in multiple myeloma therapy and the role of the UK. Brit J Haematol 191:542–551, 2020. doi:10.1111/bjh.17148
Gulla A, Anderson KC: Multiple myeloma: the (r)evolution of current therapy and a glance into the future. Haematologica 105:2358–2367, 2020. doi:10.3324/haematol.2020.247015
Premkumar V, Bhutani D, Lentzsch S: Modern treatments and future directions for relapsed/refractory multiple myeloma patients. Clinical Lymphoma Myeloma Leuk20(11):736–743, 2020. doi:10.1016/j.clml.2020.06.023
Soekojo CY, Chng WJ: Treatment horizon in multiple myeloma. Eur J Haematol 109:425-440, 2022. doi: 10.1111/ejh.13840









