



Scleritis is a severe, destructive, vision-threatening inflammation involving the deep episclera and sclera. Symptoms are moderate to marked pain, hyperemia of the globe, lacrimation, and photophobia. Diagnosis is clinical. Treatment is with systemic corticosteroids and possibly other immunosuppressants.
Scleritis is most common among women aged 30 to 50 years (1), and many also have a systemic rheumatic disease, such as rheumatoid arthritis, systemic lupus erythematosus, polyarteritis nodosa, granulomatosis with polyangiitis, or relapsing polychondritis.
A few cases are infectious in origin. About half of the cases of scleritis have no known cause.
Scleritis can affect the anterior and posterior segments of the sclera but most commonly involves the anterior segment. It can be further divided and occurs into 3 subtypes—diffuse, nodular, and necrotizing.
General reference
1. Galor A, Thorne JE. Scleritis and peripheral ulcerative keratitis. Rheum Dis Clin North Am. 2007;33(4):835-vii. doi:10.1016/j.rdc.2007.08.002
Symptoms and Signs of Scleritis
Scleritis causes pain, often characterized as a deep, boring ache, that is severe enough to interfere with sleep and appetite. Photophobia and lacrimation may occur. Hyperemic patches develop deep beneath the bulbar conjunctiva and are more violaceous than those of episcleritis or conjunctivitis. The palpebral conjunctiva is normal.
The involved area may be focal (usually one quadrant of the globe) or involve the entire globe (diffuse scleritis) and may contain a hyperemic, edematous, raised nodule (nodular scleritis) or an avascular area (necrotizing scleritis).
Intermediate scleritis and posterior scleritis are less common and are less likely to cause red eye but more likely to cause floaters, blurring, or decreased vision.

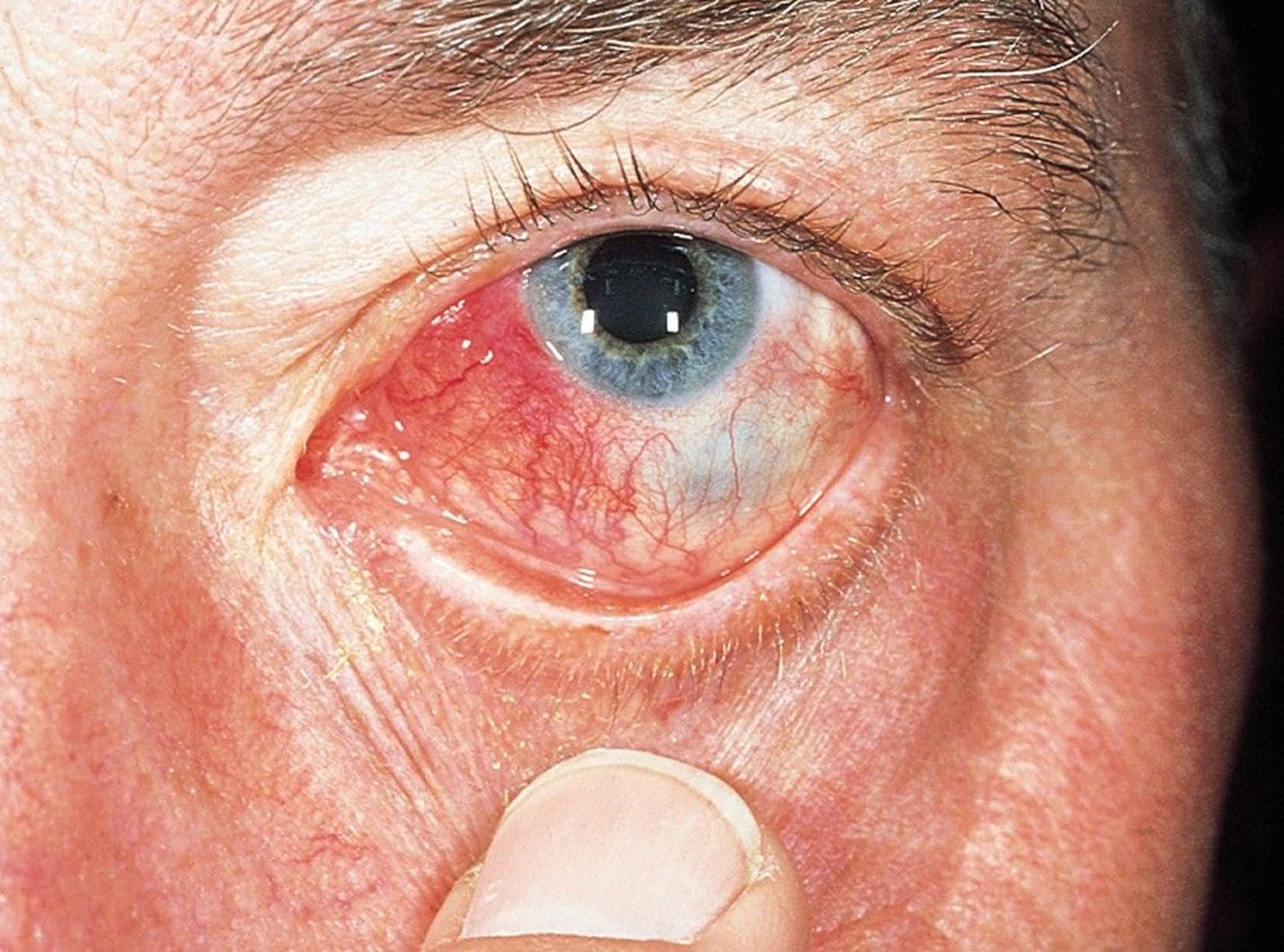
This photo shows a hyperemic, edematous, and raised nodule (nodular scleritis).
© Springer Science+Business Media

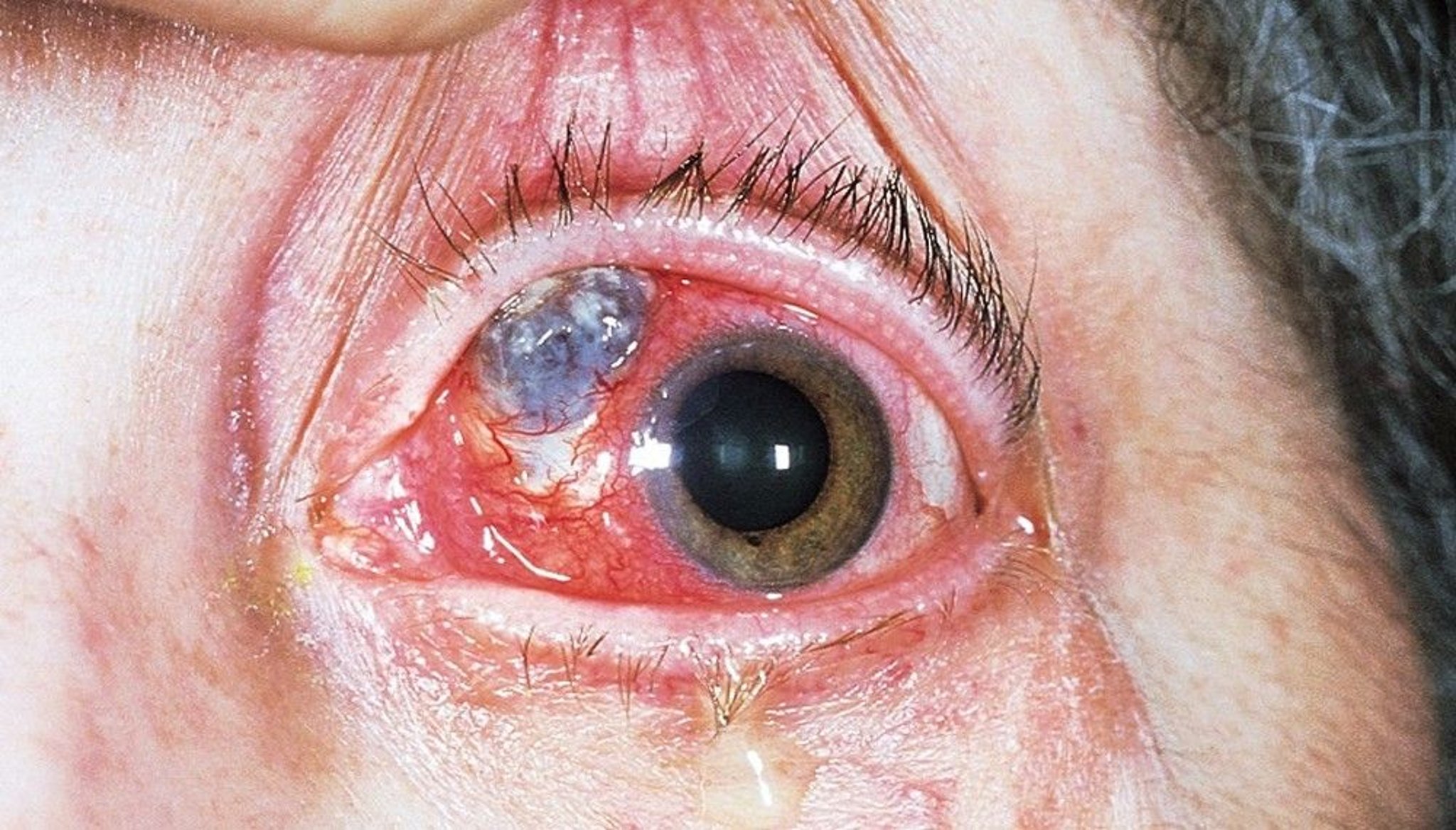
This photo shows conjunctival ulceration with scleral necrosis (necrotizing scleritis). The blue to grayish area is the choroid, visible where there has been severe scleral loss. The smaller, white, triangular area is an avascular area that has not thinned.
© Springer Science+Business Media
In severe cases of necrotizing scleritis, perforation of the globe and loss of the eye may result.
One study showed systemic rheumatic disease occurs in 8 to 25% of patients with diffuse or nodular scleritis and in 86% of patients with necrotizing scleritis (1). Necrotizing scleritis in patients with systemic rheumatic disease signals underlying systemic vasculitis.
Clinicians should carefully consider infection in patients with necrotizing scleritis because infectious scleritis may manifest similarly and because infection may complicate necrotizing scleritis.
Symptoms and signs reference
1. Bin Ismail MA, Lim RHF, Fang HM, et al. Ocular Autoimmune Systemic Inflammatory Infectious Study (OASIS)-report 4: analysis and outcome of scleritis in an East Asian population. J Ophthalmic Inflamm Infect. 2017;7(1):6. doi:10.1186/s12348-017-0124-5
Diagnosis of Scleritis
Slit-lamp examination
Diagnosis of scleritis is made clinically and by slit-lamp examination, which typically reveals a scleral violaceous hue that originates deep to the conjunctiva.
Smears or rarely biopsies are necessary to confirm infectious scleritis.
CT or ultrasound may be needed for posterior scleritis.
Treatment of Scleritis
Oral nonsteroidal anti-inflammatory drugs (NSAIDs) for mild cases
Systemic corticosteroids
Sometimes other immunosuppressants
Rarely, NSAIDs are sufficient for mild cases of scleritis. Topical corticosteroids (eg, prednisolone acetate, fluorometholone) are sometimes used in combination with NSAIDs but have limited efficacy in treating the underlying etiology of scleritis but may reduce ocular surface inflammation or congestion.
Initial therapy is usually a systemic corticosteroid (eg, prednisone 1 to 2 mg/kg orally once a day for 7 days, then tapered off by 10 mg each week as tolerated or as indicated based on the clinical picture) (1). If inflammation returns, a longer course of oral corticosteroids or pulse doses of intravenous corticosteroids (eg, methylprednisolone 1000 mg IV daily for 3 days) can be tried.
If patients have an inadequate response to low-dose corticosteroids for longer periods of time or have necrotizing scleritis and systemic rheumatic disease, systemic immunosuppression with cyclophosphamide, methotrexate, mycophenolate mofetil, or biologic agents (eg, rituximab, adalimumab) is indicated, often in consultation with a rheumatologist or qualified ophthalmologist.
Scleral grafts may be indicated for threatened perforation (2).
If infectious scleritis is suspected, broad-spectrum topical and systemic antibiotics should be administered (3, 4).
|
Treatment references
1. Daniel Diaz J, Sobol EK, Gritz DC. Treatment and management of scleral disorders. Surv Ophthalmol. 2016;61(6):702-717. doi:10.1016/j.survophthal.2016.06.002
2. Sainz de la Maza M, Tauber J, Foster CS. Scleral grafting for necrotizing scleritis. Ophthalmology. 1989;96(3):306-310. doi:10.1016/s0161-6420(89)32892-2
3. Yu J, Syed ZA, Rapuano CJ. Infectious Scleritis: Pathophysiology, Diagnosis, and Management. Eye Contact Lens. 2021;47(8):434-441. doi:10.1097/ICL.0000000000000813
4. Helm CJ, Holland GN, Webster RG Jr, Maloney RK, Mondino BJ. Combination intravenous ceftazidime and aminoglycosides in the treatment of pseudomonal scleritis. Ophthalmology. 1997;104(5):838-843. doi:10.1016/s0161-6420(97)30225-5
Prognosis for Scleritis
The prognosis for scleritis depends on the disease severity. Ocular complications are more likely to occur in patients with more severe disease (eg, necrotizing scleritis, posterior scleritis).
A 12-year longitudinal study showed 16% of patients with scleritis had a decrease in visual acuity (1).
Prognosis reference
1. Jabs DA, Mudun A, Dunn JP, Marsh MJ. Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol. 2000;130(4):469-476. doi:10.1016/s0002-9394(00)00710-8
Key Points
Scleritis is severe, destructive, vision-threatening inflammation.
Symptoms include deep, boring ache; photophobia and tearing; and focal or diffuse eye redness.
Diagnosis is made clinically and by slit-lamp examination.
Most patients require systemic corticosteroids and/or systemic immunosuppressive therapy, often prescribed in consultation with a rheumatologist.
Scleral grafts may be indicated for threatened perforation.
Drug Information for the Topic





