



The idiopathic inflammatory myopathies are characterized by inflammatory and degenerative changes in the muscles (eg, polymyositis, immune-mediated necrotizing myopathy) or in the skin and muscles (dermatomyositis). Manifestations include symmetric weakness, occasionally tenderness and myalgias, and fibrous replacement of muscle, sometimes with atrophy, principally of the proximal limb girdle muscles. Diagnosis is by clinical findings and abnormalities on laboratory, imaging, and muscle tests, which may include serum creatine kinase, MRI, electromyography, and muscle biopsy. Several types of myositis have pulmonary and cardiac manifestations, and myositis can be part of other defined systemic disorders (eg, systemic sclerosis, systemic lupus erythematosus). Treatment is with corticosteroids combined with other immunosuppressants and/or IV immune globulin.
Except for inclusion body myositis, most other forms of idiopathic inflammatory myopathy are more common among females than males (1). These disorders may appear at any age but occur most commonly from age 40 to 60 or, in children, from age 5 to 15.
General reference
1. Lilleker JB, Vencovsky J, Wang G, et al. The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann Rheum Dis. 2018;77(1):30-39. doi:10.1136/annrheumdis-2017-211868
Etiology of Idiopathic Inflammatory Myopathies
The cause of idiopathic inflammatory myopathy seems to be an autoimmune reaction to muscle tissue in genetically susceptible people. Familial clustering occurs, and human leukocyte antigen (HLA) subtypes are associated with myositis. For example, the alleles of the 8.1 ancestral haplotype (HLA-DRB1*03-DQA1*05-DQB1*02) increase risk of polymyositis, dermatomyositis, and interstitial lung disease. Possible inciting events include viral infection, certain medications, and underlying cancer. The association of cancer with dermatomyositis and necrotizing myopathies suggests that a tumor may incite myositis as the result of an autoimmune reaction against a common antigen in the muscle and in the tumor (1). Immune checkpoint inhibitors (used in the treatment of certain cancers) may induce severe myositis and sometimes myocarditis and myasthenia gravis (triple M sydrome).
Etiology reference
1. De Vooght J, Vulsteke JB, De Haes P, Bossuyt X, Lories R, De Langhe E. Anti-TIF1-γ autoantibodies: warning lights of a tumour autoantigen. Rheumatology (Oxford). 2020;59(3):469-477. doi:10.1093/rheumatology/kez572
Pathophysiology of Idiopathic Inflammatory Myopathies
Pathologic changes include cellular damage and atrophy, with variable degrees of inflammation. Muscles in the hands, feet, and face are affected less than other skeletal muscles. Involvement of muscles in the pharynx and upper esophagus and occasionally the heart can impair the functions of those organs. Inflammation may occur in joints and lungs, especially in patients with antisynthetase antibodies.
Dermatomyositis is characterized by immune complex deposition in the vessels and is considered a complement-mediated vasculopathy. In contrast, polymyositis is characterized by direct T cell–mediated muscle injury, and immune-mediated necrotizing myopathies are characterized by macrophage-predominant infiltrates and myophagocytosis (1).
Pathophysiology reference
1. Mammen A: Autoimmune muscle disease. Handb Clin Neurol. 2016;133:467–484. doi:10.1016/B978-0-444-63432-0.00025-6
Classification of Idiopathic Inflammatory Myopathies
Idiopathic inflammatory myopathies can be classified into several subgroups, mainly based on histopathology and clinical presentation (1):
Dermatomyositis (including amyopathic dermatomyositis)
Antisynthetase syndrome
Immune-mediated necrotizing myopathy
Inclusion body myositis
Polymyositis
Overlap myositis
Dermatomyositis is characterized by the presence of unique skin manifestations such as heliotrope rash, Gottron papules, and shawl and V signs that are not present in other inflammatory myopathies. Dystrophic calcification is considered a unique feature of dermatomyositis that occurs in juveniles, and, less commonly, in adults.
Presenting symptoms of antisynthetase syndrome may be a combination of various features, including seronegative inflammatory arthritis (usually nonerosive), fever, interstitial lung disease, proximal muscle weakness, hyperkeratosis of the radial aspect of the digits (mechanic's hands), and Raynaud syndrome.
Immune-mediated necrotizing myopathies most often include signal recognition particle (SRP) antibody–related myositis and statin-induced myositis. These myopathies usually have a severe presentation, often with dysphagia, respiratory muscle involvement, and very elevated creatine kinase (CK) levels, but no association with interstitial lung disease or dermatologic manifestations.
Inclusion body myositis causes proximal leg muscle weakness and frequently involves distal muscles (eg, hand and foot muscles) often with muscle wasting. It develops at an older age, has a slower progression, have lower levels of muscle enzymes compared to other inflammatory myopathies, and does not generally respond to immunosuppressive therapy.
Polymyositis is an uncommon form of inflammatory myopathy, thus the term is less frequently used. Patients with polymyositis develop myositis without skin involvement.
Inflammatory myopathy can also overlap with other systemic rheumatic diseases such as systemic lupus erythematosus and systemic sclerosis. Patients with overlap myositis have symptoms and signs of the other disorders in addition to myositis.
Classification reference
1. Lundberg IE, Fujimoto M, Vencovsky J, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers 7(1):86, 2021. doi:10.1038/s41572-021-00321-x
Symptoms and Signs of Idiopathic Inflammatory Myopathies
Onset of an inflammatory myopathy may be acute (particularly in children) or insidious (particularly in adults). Polyarthralgias, Raynaud syndrome, dysphagia, pulmonary symptoms (eg, cough, dyspnea), and constitutional complaints (notably fever, fatigue, and weight loss) may also occur. Severe disease is characterized by dysphagia, dysphonia, and/or diaphragmatic weakness.
Muscle weakness may progress over weeks to months. Patients may have difficulty raising their arms above their shoulders (eg, to brush their hair), climbing steps, or rising from a sitting position. Sometimes muscle tenderness and atrophy are present in addition to weakness. Patients may require the use of a wheelchair or become bedridden because of weakness of pelvic and shoulder girdle muscles. The flexors of the neck may be severely affected, causing an inability to raise the head from the pillow. Involvement of pharyngeal and upper esophageal muscles may impair swallowing and predispose to aspiration. Muscles of the hands, feet, eyes, and face are not involved except in inclusion body myositis, in which distal involvement, especially of the hands, is characteristic. Limb contractures rarely develop.
Joint manifestations include polyarthralgia or polyarthritis with swelling and other characteristics of nondeforming arthritis. They occur more often in a subset with Jo-1 or other antisynthetase antibodies.
Visceral involvement (except that of the pharynx and upper esophagus) is less common in idiopathic inflammatory myopathies than in some other systemic rheumatic diseases (eg, systemic lupus erythematosus, systemic sclerosis). Occasionally, and especially in patients with antisynthetase antibodies, interstitial lung disease and associated pulmonary hypertension (manifested by dyspnea and cough) are the most prominent manifestations. Cardiac involvement, especially pericarditis and cardiomyopathy, can occur. Gastrointestinal symptoms are more common in patients with an overlap syndrome with systemic sclerosis. In patients with juvenile dermatomyositis, an associated gastrointestinal vasculitis may occur and manifests as abdominal pain, hematemesis, melena, or ischemic bowel perforation.
Skin changes, which occur in dermatomyositis, tend to be dusky and erythematous and are severe when they occur with photosensitivity and skin ulceration. Periorbital edema with a purplish appearance (heliotrope rash) is relatively specific for dermatomyositis. Rash on the scalp may appear psoriaform and be intensely pruritic. Elsewhere, the rash may be slightly elevated and smooth or scaly; it may characteristically appear on the forehead, V of the neck and shoulders, chest and back, forearms and lower legs, lateral thighs, elbows and knees, medial malleoli, and dorsal aspects of the proximal interphalangeal and metacarpophalangeal joints (Gottron papules—a relatively specific finding). Cuticle overgrowth is common. Subcutaneous and muscle calcification may occur, particularly in patients with the juvenile form. The primary skin lesions frequently fade completely but may be followed by secondary changes (eg, brownish pigmentation, atrophy, persistent neovascularization, scarring).
Characteristic skin changes can occur in the absence of muscle disease (amyopathic dermatomyositis).


This photo shows skin eruption on the face of a patient with dermatomyositis. This patient has photodistributed erythema across her forehead, cheeks, and the bridge of her nose. There is violaceous erythema involving her upper eyelids and in the periorbital region consistent with a heliotrope rash.
This photo shows skin eruption on the face of a patient with dermatomyositis. This patient has photodistributed erythem
RICHARD USATINE MD / SCIENCE PHOTO LIBRARY


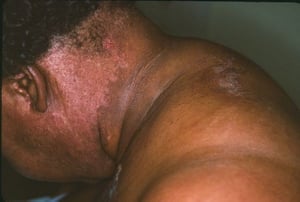
This photo shows a cutaneous manifestation dermatomyositis in a patient with colon cancer. There is a dusky, erythematous rash in a photodistributed area of the neck (V sign) that is characteristic of dermatomyositis.
This photo shows a cutaneous manifestation dermatomyositis in a patient with colon cancer. There is a dusky, erythemato
Photo courtesy of Karen McKoy, MD.

Gottron papules are scaly, erythematous to violaceous papules on extensor surfaces of proximal interphalangeal and metacarpophalangeal joints.
Gottron papules are scaly, erythematous to violaceous papules on extensor surfaces of proximal interphalangeal and meta
© Springer Science+Business Media


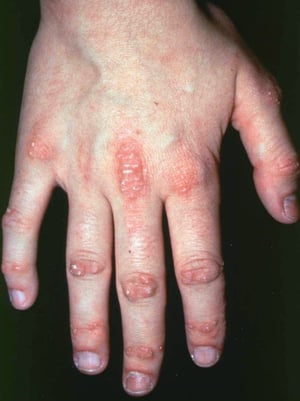
This photo shows erythematous papules over the metacarpophalangeal and interphalangeal joints and cuticle overgrowth.
This photo shows erythematous papules over the metacarpophalangeal and interphalangeal joints and cuticle overgrowth.
Photo courtesy of Kinanah Yaseen, MD.


Dermatomyositis may manifest with a dusky, erythematous rash on the dorsum of the hand, including the radiodorsal aspects of the proximal interphalangeal and metacarpophalangeal joints (Gottron papules). Dermatomyositis has an increased risk of breast, lung, ovarian, and gastrointestinal cancers in adults over age 40.
Dermatomyositis may manifest with a dusky, erythematous rash on the dorsum of the hand, including the radiodorsal aspec
Photo provided by Thomas Habif, MD.

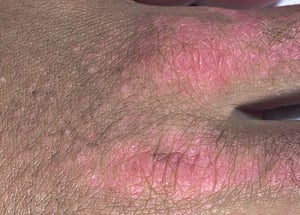
This photo shows Gottron papules (on metacarpophalangeal joints), subcutaneous calcifications (on metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints), and periungual erythema and thickening.
This photo shows Gottron papules (on metacarpophalangeal joints), subcutaneous calcifications (on metacarpophalangeal,
© Springer Science+Business Media

This photo shows dusky, erythematous skin changes resulting from dermatomyositis.
This photo shows dusky, erythematous skin changes resulting from dermatomyositis.
Photo courtesy of Karen McKoy, MD.

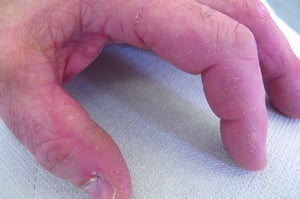
This photo shows hyperkeratosis and fissuring of the sides of the fingers in a patient with antisynthetase syndrome.
This photo shows hyperkeratosis and fissuring of the sides of the fingers in a patient with antisynthetase syndrome.
© Springer Science+Business Media


This photo shows hyperkeratotic changes of skin on the tips and sides of the fingers.
This photo shows hyperkeratotic changes of skin on the tips and sides of the fingers.
Photo courtesy of Kinanah Yaseen, MD.
This photo shows skin eruption on the face of a patient with dermatomyositis. This patient has photodistributed erythema across her forehead, cheeks, and the bridge of her nose. There is violaceous erythema involving her upper eyelids and in the periorbital region consistent with a heliotrope rash.
This photo shows skin eruption on the face of a patient with dermatomyositis. This patient has photodistributed erythem
RICHARD USATINE MD / SCIENCE PHOTO LIBRARY
This photo shows a cutaneous manifestation dermatomyositis in a patient with colon cancer. There is a dusky, erythematous rash in a photodistributed area of the neck (V sign) that is characteristic of dermatomyositis.
This photo shows a cutaneous manifestation dermatomyositis in a patient with colon cancer. There is a dusky, erythemato
Photo courtesy of Karen McKoy, MD.
Gottron papules are scaly, erythematous to violaceous papules on extensor surfaces of proximal interphalangeal and metacarpophalangeal joints.
Gottron papules are scaly, erythematous to violaceous papules on extensor surfaces of proximal interphalangeal and meta
© Springer Science+Business Media
This photo shows erythematous papules over the metacarpophalangeal and interphalangeal joints and cuticle overgrowth.
This photo shows erythematous papules over the metacarpophalangeal and interphalangeal joints and cuticle overgrowth.
Photo courtesy of Kinanah Yaseen, MD.
Dermatomyositis may manifest with a dusky, erythematous rash on the dorsum of the hand, including the radiodorsal aspects of the proximal interphalangeal and metacarpophalangeal joints (Gottron papules). Dermatomyositis has an increased risk of breast, lung, ovarian, and gastrointestinal cancers in adults over age 40.
Dermatomyositis may manifest with a dusky, erythematous rash on the dorsum of the hand, including the radiodorsal aspec
Photo provided by Thomas Habif, MD.
This photo shows Gottron papules (on metacarpophalangeal joints), subcutaneous calcifications (on metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints), and periungual erythema and thickening.
This photo shows Gottron papules (on metacarpophalangeal joints), subcutaneous calcifications (on metacarpophalangeal,
© Springer Science+Business Media
This photo shows dusky, erythematous skin changes resulting from dermatomyositis.
This photo shows dusky, erythematous skin changes resulting from dermatomyositis.
Photo courtesy of Karen McKoy, MD.
This photo shows hyperkeratosis and fissuring of the sides of the fingers in a patient with antisynthetase syndrome.
This photo shows hyperkeratosis and fissuring of the sides of the fingers in a patient with antisynthetase syndrome.
© Springer Science+Business Media
This photo shows hyperkeratotic changes of skin on the tips and sides of the fingers.
This photo shows hyperkeratotic changes of skin on the tips and sides of the fingers.
Photo courtesy of Kinanah Yaseen, MD.
Diagnosis of Idiopathic Inflammatory Myopathies
History and physical examination
Laboratory studies, electromyography, magnetic resonance imaging (MRI), and sometimes biopsy
The diagnosis of inflammatory myopathies requires a detailed clinical history and physical examination in addition to laboratory studies, electromyography, MRI, and, if available, histologic evaluation.
Laboratory studies
Laboratory studies can increase or decrease suspicion for inflammatory myopathies, assess organ involvement and its severity, and help identify overlap syndromes.
Muscle enzymes, which include creatinine kinase (CK), aldolase, and aminotransferases, may be elevated. CK is considered the most specific test; aldolase and aminotransferases are also present in hepatocytes. Skeletal muscle can also release troponin T. Elevated troponin I, however, may indicate cardiac involvement.
Myositis-specific and sometimes myositis-associated autoantibodies should be tested, some of which are available as a panel. Antinuclear antibodies (ANA) are positive in up to 80% of patients with dermatomyositis and polymyositis. If the ANA test is positive, further testing for specific types of antibodies is helpful for increasing suspicion of an overlap syndrome. However, a negative ANA test does not exclude an inflammatory myopathy.
Clinical course and manifestations are associated with particular antibodies as described in table . The relationship between these autoantibodies and disease pathogenesis remains unclear, although antibody to Jo-1 is a significant marker for pulmonary fibrosis, arthritis, and Raynaud syndrome.
Myositis-Specific and -Associated Autoantibodies
Autoantibodies | Clinical Features |
|---|---|
Antisynthetase syndrome* | |
Anti-Jo-1 | Myositis† Interstitial lung disease |
Anti-PL-7 | Interstitial lung disease† Myositis |
Anti-PL-12 | Interstitial lung disease† Myositis |
Anti-EJ | Interstitial lung disease Myositis |
Anti-OJ | Interstitial lung disease Myositis |
Anti-KS | Interstitial lung disease† Myositis |
Anti-Zo | Interstitial lung disease Myositis |
Anti-Ha | Interstitial lung disease Myositis |
Dermatomyositis | |
Anti-Mi-2 | Severe skin disease Responds well to treatment |
Anti-MDA5 | Cardiopulmonary syndrome, severe interstitial lung disease Amyopathic dermatomyositis, palmar papules, skin ulcers |
Anti-TIF1g | Increased cancer risk |
Anti-NXP-2 | Subcutaneous calcinosis Increased cancer risk |
Immune-mediated necrotizing myopathy | |
Anti-SRP | Severe, difficult to treat |
Anti-HMGCR | Immune-mediated statin-associated myopathy (can rarely be present without previous exposure to prescription statins); may persist after stopping statin therapy |
Inclusion body myositis | |
Cytosolic 5'-nucleotidase 1A | Inclusion body myositis |
Overlap syndrome | |
anti-PM-Scl | Myositis Systemic sclerosis Interstitial lung disease Mechanic's hands, calcinosis, Raynaud phenomenon |
anti-Ku | Myositis Systemic sclerosis (particularly limited cutaneous systemic sclerosis) Interstitial lung disease Arthritis, esophageal involvement, Raynaud phenomenon |
anti-U3-RNP | Myositis Systemic sclerosis (particularly diffuse cutaneous systemic sclerosis) |
* All of these autoantibodies can be present in patients with complete or incomplete forms of antisynthetase syndrome. Antisynthetase syndrome features include fever, nonerosive arthritis, interstitial lung disease, hyperkeratosis of the radial aspect of the digits (mechanic's hands), and Raynaud phenomenon. The rash of dermatomyositis may or may not be present. Patients with antisynthetase syndrome antibodies and muscle disease may have findings of dermatomyositis or polymyositis. | |
† This feature is the more prominent one. | |
Data from Mammen A: Autoimmune muscle disease. Handb Clin Neurol. 2016;133:467–484. doi:10.1016/B978-0-444-63432-0.00025-6 and from Fredi M, Cavazzana I, Franceschini F. The clinico-serological spectrum of overlap myositis. Curr Opin Rheumatol. 2018;30(6):637-643. doi:10.1097/BOR.0000000000000536. | |
Electromyography and imaging tests
Electromyography (EMG) with or without magnetic resonance imaging (MRI) of the affected muscles (eg, thighs) should be considered in patients with muscle weakness and elevated muscle enzymes. These studies help rule out neurologic causes of weakness and also identify a biopsy site.
Chest radiography, computed tomography (CT), and echocardiography are recommended when interstitial lung disease is suspected or the patient has associated pulmonary hypertension. Baseline spirometry, lung volumes, diffusion lung capacity, and respiratory muscles testing are also recommended at initial diagnosis and at follow up for patients with known lung disease.
A modified barium swallow test is needed when dysphagia is present.


This CT scan shows symmetric, bilateral lower lobe ground-glass opacities with areas of fine subpleural reticulation; mild traction bronchiectasis in both lungs with subpleural sparing; and a few scattered thoracic lymph nodes.
Photo courtesy of Kinanah Yaseen, MD.
Biopsy
Biopsy findings can vary, but chronic inflammation with muscle degeneration and some regeneration is typical.
To increase the sensitivity of biopsy results, the sample should be obtained from a muscle that has 1 or more of the following characteristics:
Weakness on physical examination
Muscle edema identified on MRI
Contralateral muscle shown to be abnormal on electromyography
Polymyositis and dermatomyositis can often be distinguished by muscle biopsy. In polymyositis, histology is usually characterized by predominant endomysial CD8+ T cell infiltrates without vasculopathy, whereas in dermatomyositis, histology is characterized by perifascicular B cell–predominant infiltrates with perivascular inflammation. A definite diagnosis made by muscle biopsy is recommended before treatment of polymyositis to exclude other muscle disorders, such as those caused by missing or defective enzymes, necrotizing myositis, and postviral rhabdomyolysis.
Muscle biopsy is not usually necessary when skin findings are characteristic of dermatomyositis. Although there is no pathognomic skin finding for dermatomyositis on biopsy, the absence of direct immunofluorescence may help distinguish skin findings from the rash in patients with systemic lupus erythematosus.
Histologic findings in inclusion body myositis include endomysial infiltrates, myofiber degeneration, and rimmed vacuoles, whereas histologic findings in immune-mediated necrotizing myopathy include prominent necrotic muscle fibers with regeneration. There is a paucity of inflammatory infiltrates, but, when present, is composed of macrophage infiltrates.


This biopsy specimen shows endomysial infiltrates, myofiber degeneration, and rimmed vacuoles (arrow).
Photo courtesy of Kinanah Yaseen, MD.
Additional testing
Cancer screening should be considered as part of the evaluation (1). Evidence for increased risk of cancer is relatively consistent in dermatomyositis but is less certain for polymyositis (2). The most commonly associated cancers in Western populations include ovarian, cervical, lung, bladder, and prostate and, in Asian populations, nasopharyngeal (3).
Cancer screening should include at least a physical examination that includes the breasts, pelvis, and rectum (with occult blood testing); complete blood count; biochemical profile; mammogram; urinalysis; chest radiograph; and any other age-appropriate cancer screening (1).
Additional investigation should be based on history and physical examination findings. Some authorities recommend CT of the neck, chest, abdomen, and pelvis as well as ovarian ultrasound, colonoscopy, and sometimes whole-body positron emission tomography (PET) in very high-risk patients (eg, patients > 40 years old with acute-onset dermatomyositis and positive myositis autoantibodies associated with cancer) (1).
Diagnosis references
1. Oldroyd AGS, Callen JP, Chinoy H, et al. International Guideline for Idiopathic Inflammatory Myopathy-Associated Cancer Screening: an International Myositis Assessment and Clinical Studies Group (IMACS) initiative [published correction appears in Nat Rev Rheumatol. 2024 May;20(5):315. doi: 10.1038/s41584-024-01111-x]. Nat Rev Rheumatol. 2023;19(12):805-817. doi:10.1038/s41584-023-01045-w
2. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291. doi:10.3899/jrheum.140566
3. Irekeola AA, Shueb RH, E A R ENS, et al. Prevalence of Nasopharyngeal Carcinoma in Patients with Dermatomyositis: A Systematic Review and Meta-Analysis. Cancers (Basel). 2021;13(8):1886. Published 2021 Apr 14. doi:10.3390/cancers13081886
Treatment of Idiopathic Inflammatory Myopathies
Corticosteroids
Other immunosuppressants (eg, methotrexate, azathioprine, mycophenolate mofetil, rituximab, tacrolimus)
IV immune globulin
Physical activities should be modestly curtailed until disease activity abates (1). Speech therapy is recommended for patients with dysphagia.
Corticosteroids are the initial medications of choice (2). The overall goal is to rapidly eliminate inflammation but minimize long-term corticosteroid exposure. For acute disease, adults receive oral prednisone approximately 1 mg/kg (up to a maximum of 80 mg) once a day. For severe disease with dysphagia or respiratory muscle weakness, treatment usually starts with intravenous high-dose corticosteroid therapy (eg, methylprednisolone 0.5 to 1 g IV once a day for 3 to 5 days), but there are no data from randomized trials to confirm that this is the optimal approach. Long-term use of corticosteroids may induce myopathy, which is usually associated with normal muscle enzymes; thus, regular examination of muscle strength during follow-up is important.
A second medication (typically methotrexate, tacrolimus, or azathioprine) is started at the same time as corticosteroids or shortly after so that prednisone can be tapered, ideally within approximately 6 months. IV immune globulin is the preferred therapy for patients who have dysphagia and respiratory muscle involvement, do not respond rapidly to corticosteroids, or develop complications with high-dose corticosteroids and other immunosuppressants, or who are also undergoing chemotherapy. Some experts may use triple therapy in severe cases or when any corticosteroid toxicity is present.Azathioprine, tacrolimus, mycophenolate mofetil, or rituximab is preferred over methotrexate in patients who have interstitial lung disease.
IV cyclophosphamide and plasma exchange can be considered for patients who have MDA-5–associated, rapidly progressive interstitial lung disease (3, 4).
Serial measurements of creatine kinase (CK) provide an early guide to therapeutic effectiveness. However, in patients with widespread muscle atrophy, levels are occasionally normal despite chronic, active myositis. MRI findings of muscle edema or high CK levels generally differentiate a relapse of myositis from corticosteroid-induced myopathy. Other muscle enzyme levels can provide clues too. Aldolase is less specific for muscle injury than CK but can occasionally be positive in patients with myositis and normal CK levels. Elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels may also reflect muscle injury.
CK levels usually fall toward or reach normal in 6 to 12 weeks during treatment, followed later by improved muscle strength. The corticosteroid dose can be gradually decreased once muscle strength normalizes. If muscle enzyme levels rise again, the corticosteroid dose is usually increased while awaiting the full effect of other corticosteroid-sparing medications.
Mycophenolate mofetil, methotrexate, and IV immune globulin are first-line therapies for cutaneous manifestations of dermatomyositis. Janus kinase (JAK) inhibitors are being explored as alternative treatment options for cutaneous manifestations of dermatomyositis (5).
If a patient treated with high-dose corticosteroids becomes increasingly weak after an initial response, corticosteroid myopathy should be considered. In these patients, CK level remains normal even though the patients are weaker.
Myositis associated with cancer is more refractory to corticosteroids. Cancer-associated myositis may remit if the tumor is successfully treated, and may recur if the tumor relapses.
People with an autoimmune disorder are at higher risk of atherosclerosis and should be closely monitored. Patients on long-term corticosteroid therapy should receive osteoporosis prophylaxis. If combination immunosuppressive therapy is used, patients should be given prophylaxis for opportunistic infections, such as Pneumocystis jirovecii (see prevention of Pneumocystis jirovecii pneumonia), and vaccines against common infections (eg, streptococcal pneumonia, influenza, COVID-19).
Treatment references
1. Dastmalchi M, Alexanderson H, Loell I, et al. Effect of physical training on the proportion of slow-twitch type I muscle fibers, a novel nonimmune-mediated mechanism for muscle impairment in polymyositis or dermatomyositis. Arthritis Rheum. 2007;57(7):1303-1310. doi:10.1002/art.22996
2. Oldroyd AGS, Lilleker JB, Amin T, et al. British Society for Rheumatology guideline on management of paediatric, adolescent and adult patients with idiopathic inflammatory myopathy. Rheumatology (Oxford). 2022;61(5):1760-1768. doi:10.1093/rheumatology/keac115
3. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy With High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol. 2020;72(3):488-498. doi:10.1002/art.41105
4. Shirakashi M, Nakashima R, Tsuji H, et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatology (Oxford). 2020;59(11):3284-3292. doi:10.1093/rheumatology/keaa123
5. Paik JJ, Lubin G, Gromatzky A, Mudd PN Jr, Ponda MP, Christopher-Stine L. Use of Janus kinase inhibitors in dermatomyositis: a systematic literature review. Clin Exp Rheumatol. 2023;41(2):348-358. doi:10.55563/clinexprheumatol/hxin6o
Prognosis for Idiopathic Inflammatory Myopathy
The prognosis for patients with idiopathic inflammatory myopathy is generally favorable; however, worse outcomes are associated with older age, the presence of certain myositis-specific autoantibodies (eg, anti-TIF1-g, NXP-2, MDA-5), and associated comorbidities (eg, severe interstitial lung disease, cancer, recurrent infection) (1, 2). For example, in a large cohort study of 628 patients with idiopathic inflammatory myopathy, the cumulative survival rate at 1 year was 91.8%, at 5 years was 82.8%, and at 10 years was 75.6% (2). However, among patients who were anti-MDA-5 positive, the 1-year survival rate was 79.5% and the 10-year survival rate was 58.5%.
Death in children with dermatomyositis may be a result of bowel vasculitis.
Prognosis references
1. Guimarães F, Yildirim R, Isenberg DA. Long-term survival of patients with idiopathic inflammatory myopathies: anatomy of a single-centre cohort. Clin Exp Rheumatol. 2023;41(2):322-329. doi:10.55563/clinexprheumatol/486yh4
2. Jiang W, Shi J, Yang H, et al. Long-Term Outcomes and Prognosis Factors in Patients With Idiopathic Inflammatory Myopathies Based on Myositis-Specific Autoantibodies: A Single Cohort Study. Arthritis Care Res (Hoboken). 2023;75(5):1175-1182. doi:10.1002/acr.24993
Key Points
Muscle weakness caused by myositis is most often proximal.
Heliotropic rash and Gottron papules are specific for dermatomyositis.
To establish the diagnosis, look for characteristic rash, muscle weakness, elevated creatine kinase level, and muscle changes on electromyography or MRI.
Unless patients have the characteristic skin findings, do a muscle biopsy to confirm the diagnosis and exclude noninflammatory myopathy.
Consider cancer screening in patients with onset of dermatomyositis at age ≥ 40 years.
Treat patients with corticosteroids and other immunosuppressants.
Drug Information for the Topic





