

Systemic lupus erythematosus is a chronic, multisystem, inflammatory disorder of autoimmune etiology, occurring predominantly in young women. Manifestations may include arthralgias and arthritis, Raynaud syndrome, malar and other rashes, pleuritis or pericarditis, renal or central nervous system involvement, and autoimmune cytopenias. Diagnosis requires clinical and serologic criteria. Treatment of severe, ongoing, active disease requires corticosteroids and immunosuppressants.
The incidence of systemic lupus erythematosus (SLE) is approximately 10-fold higher in women (usually of child-bearing age) than men (1). SLE is more common and severe among Black and Asian patients than among White patients (2, 3). It can affect patients of any age, including neonates. In some countries, the prevalence of SLE rivals that of rheumatoid arthritis.
SLE may be precipitated by currently unknown environmental triggers that cause autoimmune reactions in genetically predisposed people. Some medications (eg, hydralazine, procainamide, isoniazid, tumor necrosis factor [TNF] inhibitors) cause a reversible lupus-like syndrome.SLE may be precipitated by currently unknown environmental triggers that cause autoimmune reactions in genetically predisposed people. Some medications (eg, hydralazine, procainamide, isoniazid, tumor necrosis factor [TNF] inhibitors) cause a reversible lupus-like syndrome.

General references
1. Somers EC, Marder W, Cagnoli P, et al. Population-based incidence and prevalence of systemic lupus erythematosus: the Michigan Lupus Epidemiology and Surveillance program. Arthritis Rheumatol. 2014;66(2):369-378. doi:10.1002/art.38238
2. DeQuattro K, Trupin L, Murphy LB, et al. High Disease Severity Among Asian Patients in a US Multiethnic Cohort of Individuals With Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken). 2022;74(6):896-903. doi:10.1002/acr.24544
3. Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology (Oxford). 2017;56(11):1945-1961. doi:10.1093/rheumatology/kex260
Symptoms and Signs of SLE
Clinical findings vary greatly. SLE may develop abruptly with fever and multisystem involvement or insidiously over months or years with episodes of arthralgias and malaise. Manifestations referable to any organ system may appear. Periodic exacerbations (flares) may occur.
Joint manifestations
Joint symptoms, ranging from intermittent arthralgias to acute polyarthritis, occur in approximately 90% of patients and may precede other manifestations by years. Most lupus polyarthritis is nondestructive and nondeforming. However, in long-standing disease, deformities without bone erosions may develop (eg, the metacarpophalangeal and interphalangeal joints may rarely develop reducible ulnar drift or swan-neck deformities without bony or cartilaginous erosions [Jaccoud arthritis]) because of ligamentous laxity. Bony erosions might be detected in patients with overlap of SLE and rheumatoid arthritis (sometimes referred to as rhupus).
As in many other chronic diseases, the prevalence of fibromyalgia is increased, which may cause diagnostic confusion in patients with periarticular and generalized pain and fatigue.
Skin and mucous membrane manifestations
Skin lesions include a persistent malar butterfly erythema (flat or raised) that typically does not affect the nasolabial folds. The absence of papules and pustules and presence of skin atrophy help distinguish SLE from rosacea.
A variety of other erythematous, firm, maculopapular lesions can occur elsewhere, including exposed areas of the face and neck, upper chest, and elbows. Skin blistering and ulceration are rare, although recurrent ulcers on mucous membranes (particularly the central portion of the hard palate near the junction of the hard and soft palate, the buccal and gum mucosa, and the anterior nasal septum) are common; findings can sometimes mimic toxic epidermal necrolysis.
Generalized or focal and reversible alopecia is common during active phases of SLE. Panniculitis can cause subcutaneous nodular lesions (sometimes called lupus panniculitis or profundus). Vasculitic skin lesions may include mottled erythema on the palms and fingers, periungual erythema, nail-fold infarcts, urticaria, and palpable purpura. Petechiae may develop secondary to thrombocytopenia. Photosensitivity is common.
Lupus erythematosus tumidus is characterized by pink to violaceous nonscarring plaques and/or nodules, some annular, in light-exposed areas.
Chilblain lupus is characterized by tender, bright red to reddish blue nodules on the toes, fingers, nose, or ears that occur in cold weather. Some patients with SLE also have features of lichen planus.
Raynaud syndrome due to vasospasm in the fingers and toes causes characteristic blanching and cyanosis and might be associated with digital ischemia; however, unlike in systemic sclerosis, digital ulcers are uncommon in SLE.
(See also Cutaneous Lupus Erythematosus.)


This raised, erythematous, acute malar rash manifests in light-exposed areas (photosensitive distribution). This butterfly pattern includes the bridge of the nose, the malar areas, and the sun-exposed areas over the eyebrows. Importantly, the nasolabial folds are spared.
This raised, erythematous, acute malar rash manifests in light-exposed areas (photosensitive distribution). This butter
© Springer Science+Business Media


This photo shows discoid-like edematous lesions of the cheeks and lips that are red-brown in color.
This photo shows discoid-like edematous lesions of the cheeks and lips that are red-brown in color.
Photo courtesy of Karen McKoy, MD.


This is a photo of a patient with systemic lupus erythematosus who has discoid-like lesions of the palms and fingers with annular erythematous or hyperpigmented scaling plaques, many with central hypopigmentation.
This is a photo of a patient with systemic lupus erythematosus who has discoid-like lesions of the palms and fingers wi
Photo courtesy of Karen McKoy, MD.


This photo shows discoid lesions with hypopigmented central areas and scaly areas with surrounding erythema.
This photo shows discoid lesions with hypopigmented central areas and scaly areas with surrounding erythema.
Photo courtesy of Karen McKoy, MD.


This photo shows faintly pink edematous plaques.
This photo shows faintly pink edematous plaques.
Photo courtesy of Kinanah Yaseen, MD.
This raised, erythematous, acute malar rash manifests in light-exposed areas (photosensitive distribution). This butterfly pattern includes the bridge of the nose, the malar areas, and the sun-exposed areas over the eyebrows. Importantly, the nasolabial folds are spared.
This raised, erythematous, acute malar rash manifests in light-exposed areas (photosensitive distribution). This butter
© Springer Science+Business Media
This photo shows discoid-like edematous lesions of the cheeks and lips that are red-brown in color.
This photo shows discoid-like edematous lesions of the cheeks and lips that are red-brown in color.
Photo courtesy of Karen McKoy, MD.
This is a photo of a patient with systemic lupus erythematosus who has discoid-like lesions of the palms and fingers with annular erythematous or hyperpigmented scaling plaques, many with central hypopigmentation.
This is a photo of a patient with systemic lupus erythematosus who has discoid-like lesions of the palms and fingers wi
Photo courtesy of Karen McKoy, MD.
This photo shows discoid lesions with hypopigmented central areas and scaly areas with surrounding erythema.
This photo shows discoid lesions with hypopigmented central areas and scaly areas with surrounding erythema.
Photo courtesy of Karen McKoy, MD.
This photo shows faintly pink edematous plaques.
This photo shows faintly pink edematous plaques.
Photo courtesy of Kinanah Yaseen, MD.
Cardiopulmonary manifestations
Cardiopulmonary symptoms commonly include recurrent pleurisy, with or without pleural effusion. Pneumonitis is rare, although minor impairments in pulmonary function are common. Diffuse alveolar hemorrhage occasionally occurs and is associated with a poor prognosis. Other complications include pulmonary emboli, pulmonary hypertension, and shrinking lung syndrome.
Cardiac complications include pericarditis (most commonly) and myocarditis. Serious, rare complications are coronary artery vasculitis and valvular involvement including Libman-Sacks endocarditis. Accelerated atherosclerosis is an increasingly recognized cause of morbidity and mortality (1). Congenital heart block can develop in neonates whose mother has the antibodies against Ro (SSA) and is less common if the mother has only antibodies against La (SSB).
Lymphoid tissue
Generalized adenopathy is common, particularly among children, young adults, and African American patients. Splenomegaly can also occur.
Neurologic manifestations
Neurologic symptoms can result from involvement of any part of the central or peripheral nervous system or meninges. Mild cognitive impairment is common. There may also be headaches, personality changes, ischemic stroke, subarachnoid hemorrhage, seizures, psychoses, aseptic meningitis, peripheral and cranial neuropathies, transverse myelitis, choreoathetosis, or cerebellar dysfunction.
Differentiation between corticosteroid-induced psychosis and neuropsychiatric lupus can be challenging because neither is associated with marked abnormalities in the cerebrospinal fluid (CSF) or on routine imaging.
Renal manifestations
Renal involvement can develop at any time and may be the only manifestation of SLE (see Lupus Nephritis). It may be asymptomatic or progressive and fatal.
Renal disease can range in severity from a focal glomerulitis to a diffuse, potentially fatal membranoproliferative glomerulonephritis. Common manifestations include proteinuria (most often), an abnormal urinary sediment manifested by red blood cell casts, hypertension, and edema. Early lupus glomerulonephritis may be misdiagnosed as asymptomatic urinary tract infection.
Obstetric manifestations
Obstetric manifestations include early and late fetal loss. In patients with antiphospholipid antibodies, the risk of recurrent late miscarriages is increased. Pregnancy can be successful (see SLE in Pregnancy), particularly after 6 to 12 months of remission, but SLE flares are common during pregnancy and especially during the postpartum period. Planned pregnancy should be timed for when disease is in remission.
During pregnancy, the patient should be monitored closely for any disease flare or thrombotic events by a multidisciplinary team that includes an obstetrician who specializes in high-risk pregnancies. Women who are SSA antibody-positive should have weekly fetal ultrasonography between week 18 and week 26 to assess for congenital heart block.
Hematologic manifestations
Hematologic manifestations include anemia (anemia of chronic disease, autoimmune hemolytic anemia), leukopenia (usually lymphopenia, neutropenia, or both), and thrombocytopenia (usually mild but sometimes life-threatening autoimmune thrombocytopenia). Recurrent arterial or venous thrombosis, thrombocytopenia, and a high probability of obstetric complications occur in patients with antiphospholipid antibodies. Thromboses account for some of the complications of SLE, including obstetric complications.
Macrophage activation syndrome is a rare but potentially life-threatening complication that can occur.
Gastrointestinal manifestations
Gastrointestinal manifestations can result from bowel vasculitis or impaired bowel motility. In addition, pancreatitis can rarely result from SLE.
Manifestations may include abdominal pain resulting from serositis, nausea, vomiting, manifestations of bowel perforation, protein-losing enteropathy, and pseudo-obstruction.
SLE rarely causes parenchymal liver disease.
Symptoms and signs reference
1. Bello N, Meyers KJ, Workman J, Hartley L, McMahon M. Cardiovascular events and risk in patients with systemic lupus erythematosus: Systematic literature review and meta-analysis. Lupus. 2023;32(3):325-341. doi:10.1177/09612033221147471
Diagnosis of SLE
Clinical criteria
Cytopenias
Autoantibodies
SLE should be suspected in patients, particularly young women, with any of the symptoms and signs. However, early-stage SLE can mimic other systemic rheumatic diseases, including rheumatoid arthritis if joint symptoms predominate. Mixed connective tissue disease includes, by definition, features of SLE as well as possibly features of systemic sclerosis, rheumatoid-like polyarthritis, and myositis. Infections (eg, bacterial endocarditis, histoplasmosis) can mimic SLE and may develop as a result of treatment-caused immunosuppression. Disorders such as sarcoidosis and paraneoplastic syndromes can also mimic SLE.
Laboratory testing may differentiate SLE from other systemic rheumatic diseases. Initial laboratory testing should include the following:
Antinuclear antibodies (ANA)
Extractable nuclear antigens (ENAs) if ANA test is positive, including anti–double-stranded (ds) DNA (anti-dsDNA), anti-Smith, anti-U1 RNP, anti-Ro/SSA, and anti-La/SSB antibodies
Complement C3 and C4 levels
Complete blood count (CBC)
Urinalysis with urinary sediment
Chemistry profile including renal and liver enzymes
In clinical practice, some clinicians rely on the classification criteria for SLE developed by the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR; see table ). Patients are eligible for these criteria only if they have a positive ANA result ≥ 1:80. The 2019 EULAR/ACR classification criteria include clinical and immunologic domains, and each criterion is assigned a weight of 2 to 10. If the patient's score is 10 or more, and at least 1 clinical criterion is fulfilled, disease is classified as SLE. However, a positive ANA does not indicate a diagnosis of lupus. A positive ANA test in the presence of fatigue and generalized myofascial pain without other clinical or laboratory findings is rarely significant (1).
EULAR/ACR Criteria for the Classification of Systemic Lupus Erythematosus[a]
Domain[b] | Weight[c] |
|---|---|
Clinical domains | |
Constitutional:
|
|
Hematologic:
|
|
Neuropsychiatric:
|
|
Mucocutaneous:
|
|
Serosal:
|
|
Musculoskeletal:
|
|
Renal:
|
|
Immunologic domains | |
Antiphospholipid antibodies:
|
|
Complement proteins:
|
|
SLE-specific antibodies:
|
|
[a] Patients are eligible for these criteria only if they have a positive ANA test ≥ 1:80. | |
[b] Criteria do not need to occur simultaneously. Only the highest-weighted criterion score within a single domain should be used. SLE must be the most likely explanation for each criterion. | |
[c] Each criterion is assigned a weight of 2 to 10. If the patient's score is 10 or more, and at least 1 clinical criterion is fulfilled, disease is classified as SLE. | |
[d] Evidence of autoimmune hemolysis (such as the presence of reticulocytosis, low haptoglobin, elevated indirect bilirubin, elevated lactate dehydrogenase) and a positive direct antiglobulin (direct Coombs) test. | |
[e] This criterion may be noted during physical examination or review of a photo. | |
[f] Joint involvement is defined as either synovitis involving ≥ 2 joints characterized by swelling or effusion or tenderness in ≥ 2 joints and at least 30 minutes of morning stiffness. | |
ANA = antinuclear antibodies; anti-dsDNA = anti–double-stranded (ds) DNA; EULAR/ACR = European League Against Rheumatism/American College of Rheumatology; SLE = systemic lupus erythematosus. | |
Data from Aringer M, Costenbader K, Daikh D, et al: 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol 71(9):1400–1412, 2019. doi: 10.1002/art.40930 | |
ANA testing
Testing for ANA (preferably by indirect immunofluorescence rather than by a solid-phase assay) is an appropriate initial test for patients with suspected SLE; a positive ANA test (usually in high titer: > 1:80) occurs in > 95% of people with SLE (2). However, a positive ANA test can also occur in rheumatoid arthritis, other systemic rheumatic diseases, autoimmune thyroid disease, multiple sclerosis, cancers, and even in the general population. The false-positive rate varies from approximately 3% with ANA titers of 1:320 to approximately 30% for ANA titers of 1:40 among healthy controls. Medications such as hydralazine, procainamide, and tumor necrosis factor inhibitors can cause positive ANA results as well as a drug-induced lupus; the symptoms typically resolve after the medication is stopped. Positive ANA should prompt more specific testing such as anti-dsDNA antibodies; anti-dsDNA is highly specific for SLE (3).
Other ANA and anticytoplasmic antibodies
The ANA test is very sensitive, but it is not specific for SLE; thus, evidence of other autoantibodies is used to aid in diagnosis. The other autoantibodies are often referred to as extractable nuclear antigens and include dsDNA, Smith (Sm), ribonucleoprotein (RNP), Ro (SSA), and La (SSB).
Ro is predominantly cytoplasmic; anti-Ro antibodies are occasionally present in patients with ANA-negative SLE presenting with subacute cutaneous lupus erythematosus. Anti-Ro is the causal antibody for neonatal lupus and congenital heart block.
Anti-Sm is highly specific for SLE but, like anti-dsDNA, is not sensitive.
Anti-RNP occurs in patients with SLE, mixed connective tissue disease, and occasionally other systemic rheumatic disorders and systemic sclerosis.
Other tests
Leukopenia (usually lymphopenia and neutropenia) is common. Hemolytic anemia may occur, but low hemoglobin and red blood cell counts are more often due to the anemia of chronic disease. Thrombocytopenia in SLE may be difficult or impossible to differentiate from idiopathic thrombocytopenic purpura except that patients have other features of SLE and/or SLE-specific antibodies (anti-dsDNA or anti-Sm). False-positive serologic tests for syphilis occur in 5 to 10% of patients with SLE. These test results may be associated with the lupus anticoagulant and a prolonged partial thromboplastin time (PTT). Abnormal values in 1 or more of these assays suggest the presence of antiphospholipid antibodies (eg, anticardiolipin antibodies), which should then be measured directly by enzyme-linked immunosorbent assay (ELISA). Antiphospholipid antibodies are associated with arterial or venous thrombosis, mild thrombocytopenia, and, during pregnancy, spontaneous abortion or late fetal death but may be present in asymptomatic patients.
Other blood tests help monitor disease severity and determine the need for treatment. Serum complement levels (C3, C4) are often depressed in active disease and are usually lowest in patients with active nephritis. Erythrocyte sedimentation rate (ESR) is elevated frequently during active disease. C-reactive protein levels are not necessarily elevated; high levels raise the concern for infection and/or serositis.
Complete spirometry tests and an electrocardiogram are recommended in patients with respiratory symptoms.
Renal involvement
Screening for renal involvement begins with urinalysis with urinary sediment. Red blood cell (RBC) and/or white blood cell casts suggest active nephritis. Urinalysis should be done at regular intervals (eg, every 3 to 6 months), even for patients in apparent remission and without previous renal involvement, because kidney disease is usually asymptomatic. Proteinuria can be estimated by the urine protein/creatinine ratio or measured in a 24-hour urine collection.
Renal biopsy is indicated in patients whose protein excretion is > 500 mg/day and who have hematuria (thought to be glomerular) or RBC casts and is helpful in evaluating the status of renal disease (ie, active inflammation vs chronic changes) and in guiding therapy. Classification of lupus nephritis is based on histologic findings on renal biopsy (see table ). Repeat renal biopsy should be considered in some patients because switching from one class of lupus nephritis to another is common in patients with SLE.
Patients with chronic renal insufficiency and mostly sclerotic glomeruli are not likely to benefit from aggressive immunosuppressive therapy.
Diagnosis references
1. Nashi RA, Shmerling RH. Antinuclear Antibody Testing for the Diagnosis of Systemic Lupus Erythematosus. Med Clin North Am. 2021;105(2):387-396. doi:10.1016/j.mcna.2020.10.003
2. Tan EM, Feltkamp TE, Smolen JS, et al. Range of antinuclear antibodies in "healthy" individuals. Arthritis Rheum. 1997;40(9):1601-1611. doi:10.1002/art.1780400909
3. Kavanaugh AF, Solomon DH; American College of Rheumatology Ad Hoc Committee on Immunologic Testing Guidelines. Guidelines for immunologic laboratory testing in the rheumatic diseases: anti-DNA antibody tests. Arthritis Rheum. 2002;47(5):546-555. doi:10.1002/art.10558
Treatment of SLE
Hydroxychloroquine (an antimalarial) for all patients with SLEHydroxychloroquine (an antimalarial) for all patients with SLE
Nonsteroidal anti-inflammatory drugs (NSAIDs) as needed in addition to antimalarials for mild disease
Corticosteroids, other immunosuppressants, and antimalarials for severe disease
To guide therapy, SLE should be classified as mild to moderate (eg, fever, arthritis, pleurisy, pericarditis, rash) or severe (eg, hemolytic anemia, severe thrombocytopenic purpura, massive pleural and pericardial involvement, diffuse alveolar hemorrhage or pneumonitis, nephritis, acute vasculitis of the extremities or gastrointestinal tract, florid central nervous system [CNS] involvement).
The antimalarial hydroxychloroquine is indicated for all patients with SLE regardless of disease severity because it decreases disease flares and decreases mortality (The antimalarial hydroxychloroquine is indicated for all patients with SLE regardless of disease severity because it decreases disease flares and decreases mortality (1, 2). Hydroxychloroquine may also reduce thrombotic events especially in patients with associated antiphospholipid syndrome. It must be avoided if there is an absolute contraindication because of adverse events (eg, ocular toxicity). In addition, it should be used with caution if there is a history of glucose-6-phosphate dehydrogenase (G6PD) deficiency (3).
Patients require routine monitoring during treatment to assess disease activity and response to therapy. In addition to clinical follow-up, disease activity can be assessed with the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) (4) and the British Isles Lupus Assessment Group (BILAG) index (5).
Mild to moderate disease
Arthralgias are usually controlled with NSAIDs. However, chronic NSAID use is discouraged because of gastrointestinal adverse effects (eg, peptic ulcer disease) and potential coronary and renal toxicity (eg, interstitial nephritis, papillary necrosis). Topical agents (eg, corticosteroids, tacrolimus) can be used for skin disease, usually under the guidance of dermatology. Arthralgias are usually controlled with NSAIDs. However, chronic NSAID use is discouraged because of gastrointestinal adverse effects (eg, peptic ulcer disease) and potential coronary and renal toxicity (eg, interstitial nephritis, papillary necrosis). Topical agents (eg, corticosteroids, tacrolimus) can be used for skin disease, usually under the guidance of dermatology.
Antimalarials, such as hydroxychloroquine, are useful for joint and skin manifestations. Antimalarials, such as hydroxychloroquine, are useful for joint and skin manifestations.Hydroxychloroquine reduces the frequency of SLE flares and decreases mortality, and is therefore used in virtually all patients with SLE. The dose is 5 mg/kg of actual body weight orally once a day with a maximum dose of 400 mg/day. Baseline ophthalmologic examination should be done before starting therapy to exclude retinopathy because chronic hydroxychloroquine use increases the risk of toxic retinopathy. Ophthalmologic screening should be done yearly to assess for retinal toxicity. Hydroxychloroquine can rarely cause skeletal or cardiac muscle toxicity. Alternatives include oral chloroquine 250 mg once a day and oral quinacrine 50 to 100 mg once a day (can rarely cause skeletal or cardiac muscle toxicity. Alternatives include oral chloroquine 250 mg once a day and oral quinacrine 50 to 100 mg once a day (6).
Methotrexate (15 to 20 mg orally or subcutaneously once a week), azathioprine (2 mg/kg orally once a day), or mycophenolate mofetil (1 to 1.5 grams orally twice a day) can be added to Methotrexate (15 to 20 mg orally or subcutaneously once a week), azathioprine (2 mg/kg orally once a day), or mycophenolate mofetil (1 to 1.5 grams orally twice a day) can be added tohydroxychloroquine in patients with uncontrolled mild to moderate disease who would otherwise be candidates for a course of corticosteroids. The ultimate goal is to maintain disease remission either without the need for corticosteroids or with only the lowest dose possible.
Belimumab (10 mg/kg IV every 2 weeks for 3 doses, then 10 mg/kg IV once a month or 200 mg subcutaneously once a week) should be considered if patients have uncontrolled disease or frequent flares, particularly for joint, skin, renal, or nonsevere hematologic manifestations (Belimumab (10 mg/kg IV every 2 weeks for 3 doses, then 10 mg/kg IV once a month or 200 mg subcutaneously once a week) should be considered if patients have uncontrolled disease or frequent flares, particularly for joint, skin, renal, or nonsevere hematologic manifestations (2). It can be used in addition to hydroxychloroquine and in combination with other medications depending on the specific system involved and severity of disease. Screening and monitoring for depression is required when initiating therapy with ). It can be used in addition to hydroxychloroquine and in combination with other medications depending on the specific system involved and severity of disease. Screening and monitoring for depression is required when initiating therapy withbelimumab because of a possible risk of new-onset or worsening depression and suicidality.
Severe disease
Treatment includes induction therapy to control acute severe manifestations followed by maintenance therapy. Corticosteroids are first-line therapy. A combination of a corticosteroid and other immunosuppressants is typically used in active severe disease (ie, lupus nephritis with impaired renal function, myocarditis, or CNS involvement).
The complication for which there is the strongest evidence for treatment efficacy is lupus nephritis. Methylprednisolone 1 g by slow (1-hour) IV infusion on 3 successive days is often the initial treatment, although trial evidence for this pulse corticosteroid therapy is lacking. Then, oral prednisone given in doses of 0.5 to 1 mg/kg once a day (usually 40 to 60 mg once a day) is initiated and the dose is adjusted according to the manifestation of SLE. Corticosteroids should be tapered as soon as allowed by the disease, usually within 6 months, to limit adverse effects. Cyclophosphamide (see table . Methylprednisolone 1 g by slow (1-hour) IV infusion on 3 successive days is often the initial treatment, although trial evidence for this pulse corticosteroid therapy is lacking. Then, oral prednisone given in doses of 0.5 to 1 mg/kg once a day (usually 40 to 60 mg once a day) is initiated and the dose is adjusted according to the manifestation of SLE. Corticosteroids should be tapered as soon as allowed by the disease, usually within 6 months, to limit adverse effects. Cyclophosphamide (see table) or mycophenolate mofetil (up to 3 g a day orally in 2 doses) is also used for induction therapy along with corticosteroids. Effective birth control (an intrauterine device is typically preferred to hormonal approaches) is required when using ) or mycophenolate mofetil (up to 3 g a day orally in 2 doses) is also used for induction therapy along with corticosteroids. Effective birth control (an intrauterine device is typically preferred to hormonal approaches) is required when usingmycophenolate mofetil and cyclophosphamide because of the risk of congenital malformations.
Adding belimumab in a dose of 10 mg/kg IV monthly to corticosteroids and mycophenolate or corticosteroids and cyclophosphamide has been shown to lead to a better renal response and complete renal response at 6 months compared to corticosteroids and mycophenolate or corticosteroids and Adding belimumab in a dose of 10 mg/kg IV monthly to corticosteroids and mycophenolate or corticosteroids and cyclophosphamide has been shown to lead to a better renal response and complete renal response at 6 months compared to corticosteroids and mycophenolate or corticosteroids andcyclophosphamide alone, especially if extrarenal manifestations are active (7). Voclosporin in a dose of 23.7 mg orally twice a day in combination with ). Voclosporin in a dose of 23.7 mg orally twice a day in combination withmycophenolate mofetil and a rapidly tapered course of corticosteroid has been shown to lead to better renal outcomes at 1 year than corticosteroids and mycophenolate mofetil alone (8). Both belimumab and voclosporin are now often being used in combination with mycophenolate to treat lupus nephritis (classes III, IV, and V), but clear guidelines for their use are not yet available (9).
Cyclophosphamide use for more than 6 months is discouraged because of potential toxicities, including infertility and increased risk of cancer. Once disease control is achieved, patients are transitioned to either mycophenolate mofetil (1 to 1.5 g orally 2 times a day) or azathioprine (0.5 to 1.5 mg/kg orally twice a day) for maintenance. Women of childbearing age for whom Cyclophosphamide use for more than 6 months is discouraged because of potential toxicities, including infertility and increased risk of cancer. Once disease control is achieved, patients are transitioned to either mycophenolate mofetil (1 to 1.5 g orally 2 times a day) or azathioprine (0.5 to 1.5 mg/kg orally twice a day) for maintenance. Women of childbearing age for whomcyclophosphamide is being considered should be informed about the risk of gonadal toxicity and offered a fertility consult for ovarian protection or egg harvesting when possible.
IV Cyclophosphamide Protocols for Systemic Lupus Erythematosus
Disease Complication | Regimen[a] |
|---|---|
Lupus nephritis[b] | Induction therapy for lupus nephritis: 500 mg on weeks 0, 2, 4, 6, 8, and 10[c] |
Organ- or life-threatening disease | 0.75 to 1 g/m2 BSA/month for 6 months[d,e] |
[a] Cyclophosphamide is given with a corticosteroid (eg, methylprednisolone 1 g IV every day for 3 days followed by prednisone 40 to 60 mg orally per day) and mesna. [a] Cyclophosphamide is given with a corticosteroid (eg, methylprednisolone 1 g IV every day for 3 days followed by prednisone 40 to 60 mg orally per day) and mesna.Mesna is given in a dose equal to cyclophosphamide. Mesna binds acrolein, which is a metabolite of cyclophosphamide that irritates the bladder. | |
[b] See also table Classification of Lupus Nephritis. | |
[c] Euro-Lupus regimen. | |
[d] Cyclophosphamide should not be used after this period. | |
[e] National Institutes of Health regimen. | |
BSA = body surface area. | |
Adapted from Fanouriakis A, Kostopoulou M, Alunno A, et al: 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis 78(6):736–745, 2019. doi: 10.1136/annrheumdis-2019-215089 | |
In neuropsychiatric lupus, including transverse myelitis, treatment recommendations are based on anecdotal evidence, and options include IV cyclophosphamide or IV rituximab (eg, 1 g on day 1 and day 15 given at 6-month intervals) in addition to a corticosteroid. , treatment recommendations are based on anecdotal evidence, and options include IV cyclophosphamide or IV rituximab (eg, 1 g on day 1 and day 15 given at 6-month intervals) in addition to a corticosteroid.
First-line therapy for thrombocytopenia and hemolytic anemia includes moderate- or high-dose corticosteroids (typically prednisone 1 mg/kg orally once a day, maximum 80 mg a day) along with an immunosuppressant (azathioprine 2 mg/kg orally once a day or mycophenolate mofetil 1 g orally every 12 hours). IV immune globulin 400 mg/kg once a day for 5 consecutive days or 1 g/kg once a day for 2 days may be useful, particularly if high-dose corticosteroids are contraindicated (eg, in patients with active infection). First-line therapy for thrombocytopenia and hemolytic anemia includes moderate- or high-dose corticosteroids (typically prednisone 1 mg/kg orally once a day, maximum 80 mg a day) along with an immunosuppressant (azathioprine 2 mg/kg orally once a day or mycophenolate mofetil 1 g orally every 12 hours). IV immune globulin 400 mg/kg once a day for 5 consecutive days or 1 g/kg once a day for 2 days may be useful, particularly if high-dose corticosteroids are contraindicated (eg, in patients with active infection).Rituximab is an alternative option for refractory cases (2).
Patients with end-stage renal disease can undergo kidney transplantation, as an alternative to dialysis, with a successful outcome, especially if their disease has been in remission.
Improvement of severe SLE often takes 4 to 12 weeks. Thrombosis or embolism of cerebral, pulmonary, or placental vessels requires short-term treatment with heparin and longer treatment with warfarin. If the diagnosis of antiphospholipid syndrome is confirmed, lifelong therapy (usually Improvement of severe SLE often takes 4 to 12 weeks. Thrombosis or embolism of cerebral, pulmonary, or placental vessels requires short-term treatment with heparin and longer treatment with warfarin. If the diagnosis of antiphospholipid syndrome is confirmed, lifelong therapy (usuallywarfarin) is usually indicated. The initial target international normalized ratio is usually 2 to 3.
Anifrolumab (IgG1κ monoclonal antibody to type I interferon receptor), at a dose of 300 mg IV every 4 weeks, may be added to standard therapy for the management of moderate to severe SLE, particularly in patients with severe skin disease. However, patients with active and severe neuropsychiatric or renal disease were not included in the pivotal trial (Anifrolumab (IgG1κ monoclonal antibody to type I interferon receptor), at a dose of 300 mg IV every 4 weeks, may be added to standard therapy for the management of moderate to severe SLE, particularly in patients with severe skin disease. However, patients with active and severe neuropsychiatric or renal disease were not included in the pivotal trial (10).
Use of CD19 chimeric antigen receptor (CAR) T-cell therapy for the treatment of refractory SLE shows promise (11).
Maintenance therapy
Chronic disease should be treated with the lowest dose of corticosteroids (eg, oral prednisone ≤ 7.5 mg once a day or its equivalent) and other medications that control inflammation (eg, antimalarials, immunosuppressants [mycophenolate mofetil or azathioprine]) to maintain remission (Chronic disease should be treated with the lowest dose of corticosteroids (eg, oral prednisone ≤ 7.5 mg once a day or its equivalent) and other medications that control inflammation (eg, antimalarials, immunosuppressants [mycophenolate mofetil or azathioprine]) to maintain remission (2). Treatment should be guided by clinical features primarily, although anti-dsDNA antibody titers or serum complement levels may be followed, particularly if they have correlated with disease activity in the past. However, anti-dsDNA antibody titers or serum complement levels may not parallel nonrenal disease flares. Other pertinent blood and urine tests may be used to assess specific organ involvement.
Calcium, vitamin D, and bisphosphonate therapy (see Calcium, vitamin D, and bisphosphonate therapy (seeprevention of osteoporosis) should be considered in patients taking corticosteroids long term.
If combination immunosuppressive therapy is used, patients should be given prophylaxis for opportunistic infections, such as Pneumocystis jirovecii (see prevention of Pneumocystis jirovecii pneumonia), and vaccines against common infections (eg, streptococcal pneumonia, human papillomavirus, influenza, COVID-19).
Photoprotection is also an important measure to help prevent flares. Sunscreens with a sun protection factor (SPF) > 50 that block both UVA and UVB are recommended.
Coexisting medical conditions and pregnancy
All patients should be closely monitored for atherosclerosis, and cardiovascular risk reduction is a key part of management (see treatment of atherosclerosis). Long-term anticoagulation is vital in patients who also have antiphospholipid syndrome and history of thrombosis (see also Anticoagulants).
Pregnant women should remain on hydroxychloroquine throughout their pregnancy, and low-dose aspirin is recommended as well. When clinical antiphospholipid syndrome is present, as manifested by prior thrombotic events, full anticoagulation therapy with low molecular weight or unfractionated heparin is advised. If the pregnant woman has positive antiphospholipid syndrome antibodies and prior late-stage fetal loss or recurrent first trimester miscarriages, prophylactic low molecular weight or unfractionated Pregnant women should remain on hydroxychloroquine throughout their pregnancy, and low-dose aspirin is recommended as well. When clinical antiphospholipid syndrome is present, as manifested by prior thrombotic events, full anticoagulation therapy with low molecular weight or unfractionated heparin is advised. If the pregnant woman has positive antiphospholipid syndrome antibodies and prior late-stage fetal loss or recurrent first trimester miscarriages, prophylactic low molecular weight or unfractionatedheparin can be considered during pregnancy and 6 weeks postpartum. When the patient has positive serologies but no prior obstetric or thrombotic events, recommendations are less clear. Co-management by a hematologist, an obstetrician who specializes in high-risk pregnancies, and a rheumatologist should be considered when managing these patients.
Mycophenolate mofetil is teratogenic. Because of this teratogenicity and the known poor outcomes related to active SLE during pregnancy, women should ideally conceive after their disease has been in remission for 6 months or longer. If the patient needs to remain on immunosuppression (eg, ongoing maintenance therapy for lupus nephritis), Mycophenolate mofetil is teratogenic. Because of this teratogenicity and the known poor outcomes related to active SLE during pregnancy, women should ideally conceive after their disease has been in remission for 6 months or longer. If the patient needs to remain on immunosuppression (eg, ongoing maintenance therapy for lupus nephritis),mycophenolate mofetil is usually switched to azathioprine at least 6 months prior to conceiving. Azathioprine and tacrolimus are considered safe during pregnancy.at least 6 months prior to conceiving. Azathioprine and tacrolimus are considered safe during pregnancy.
The choice of contraception method usually is based on multiple factors, including disease activity, risk of thrombosis, and patient preference (12).
Treatment references
1. Alarcón GS, McGwin G, Bertoli AM, et al. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: Data from LUMINA, a multiethnic US cohort (LUMINA L). Ann Rheum Dis. 2007;66(9):1168–1172. doi:10.1136/ard.2006.068676
2. Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78(6):736-745. doi:10.1136/annrheumdis-2019-215089
3. Kane M. Hydroxychloroquine Therapy and G6PD Genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kattman BL, Malheiro AJ, eds. Medical Genetics Summaries. Bethesda (MD): National Center for Biotechnology Information (US); May 2, 2023.
4. Touma Z, Urowitz MB, Gladman DD. SLEDAI-2K for a 30-day window. Lupus. 2010;19(1):49-51. doi:10.1177/0961203309346505
5. Romero-Diaz J, Isenberg D, Ramsey-Goldman R. Measures of adult systemic lupus erythematosus: updated version of British Isles Lupus Assessment Group (BILAG 2004), European Consensus Lupus Activity Measurements (ECLAM), Systemic Lupus Activity Measure, Revised (SLAM-R), Systemic Lupus Activity Questionnaire for Population Studies (SLAQ), Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), and Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index (SDI). Arthritis Care Res (Hoboken). 2011;63 Suppl 11(0 11):S37-S46. doi:10.1002/acr.20572
6. Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy (2016 Revision). Ophthalmology. 2016;123(6):1386-1394. doi:10.1016/j.ophtha.2016.01.058
7. Furie R, Rovin BH, Houssiau F, et al: Two-year, randomized, controlled trial of belimumab in lupus nephritis. N Engl J Med 383(12):1117-1128, 2020. doi:10.1056/NEJMoa2001180
8. Rovin BH, Teng YKO, Ginzler EM, et al: Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial.Lancet 397(10289):2070-2080, 2021. doi:10.1016/S0140-6736(21)00578-X. Erratum in: Lancet 397(10289):2048, 2021.
9. Bajema IM, Wilhelmus S, Alpers CE, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int 93(4):789-796, 2018. doi:10.1016/j.kint.2017.11.023
10. Morand EF, Furie R, Tanaka Y, et al: Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med 382(3):211-221, 2020. doi:10.1056/NEJMoa1912196
11. Müller F, Taubmann J, Bucci L, et al. CD19 CAR T-Cell Therapy in Autoimmune Disease - A Case Series with Follow-up. N Engl J Med. 2024;390(8):687-700. doi:10.1056/NEJMoa2308917
12. Sammaritano LR. Contraception in patients with systemic lupus erythematosus and antiphospholipid syndrome. Lupus. 2014;23(12):1242-1245. doi:10.1177/0961203314528062
Prognosis for SLE
The course is usually chronic, relapsing, and unpredictable. Remissions may last for years. If the initial acute phase is controlled, even if very severe (eg, with cerebral thrombosis or severe nephritis), the long-term prognosis is usually good.
The 10-year survival in most high-resource countries is almost 90% (1). Improved prognosis is in part due to earlier diagnosis and more effective therapies. However, despite advanced therapy and improvement of the mortality rate, survival is still considered lower than general population because of premature cardiovascular disease, disease activity, end-stage renal disease, and infection (2).
Prognosis references
1. Tektonidou MG, Lewandowski LB, Hu J, Dasgupta A, Ward MM. Survival in adults and children with systemic lupus erythematosus: a systematic review and Bayesian meta-analysis of studies from 1950 to 2016 [published correction appears in Ann Rheum Dis. 2018 Mar;77(3):472. doi: 10.1136/annrheumdis-2017-211663corr1]. Ann Rheum Dis. 2017;76(12):2009-2016. doi:10.1136/annrheumdis-2017-211663
2. Yen EY, Shaheen M, Woo JMP, et al. 46-Year Trends in Systemic Lupus Erythematosus Mortality in the United States, 1968 to 2013: A Nationwide Population-Based Study. Ann Intern Med. 2017;167(11):777-785. doi:10.7326/M17-0102
Key Points
Joint and skin manifestations are common in SLE, but the disorder can also affect various organ systems, such as the heart, lungs, lymphoid tissue, and kidneys and the gastrointestinal, hematologic, reproductive, and nervous systems.
The European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) criteria can be used to support the diagnosis of SLE.
Among tests, use the highly sensitive ANA for screening, but use clinical findings and other laboratory tests (eg, anti-dsDNA, anti-Sm) to help support the diagnosis.
Evaluate all patients for kidney involvement.
Treat all patients with hydroxychloroquine and, for mild disease, NSAIDs as needed.Treat all patients with hydroxychloroquine and, for mild disease, NSAIDs as needed.
Use corticosteroids for moderate or severe SLE and an additional immunosuppressant for active lupus nephritis, neuropsychiatric lupus, skin manifestations that do not respond to hydroxychloroquine, diffuse alveolar hemorrhage, vasculitis, recurrent serositis, or cardiac manifestations.
Use corticosteroids at the lowest possible dose and use other medications to maintain remission.
More Information
The following English-language resources may be useful. Please note that The Manual is not responsible for the content of these resources.
American Academy of Ophthalmology: Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy (2016 Revision)
European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR): 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus
Cutaneous Lupus Erythematosus
Discoid lupus erythematosus (DLE)
DLE is a type of chronic cutaneous lupus erythematosus. It is characterized by a set of skin changes that can occur on its own or as part of systemic lupus erythematosus (SLE).
Skin lesions begin as erythematous plaques and progress to atrophic scars. They cluster in light-exposed areas of the skin, such as the face, scalp, and ears. Untreated, lesions extend and develop central atrophy and scarring. There may be widespread scarring alopecia. Mucous membrane involvement may be prominent, especially in the mouth. Sometimes lesions are hypertrophic and may mimic lichen planus (called hypertrophic or verrucous lupus).

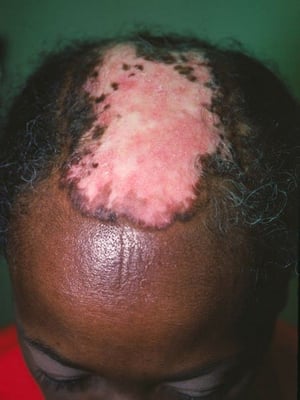
This photo shows discoid erythematous plaques causing atrophic scarring and resultant alopecia on the scalp.
This photo shows discoid erythematous plaques causing atrophic scarring and resultant alopecia on the scalp.
Photo courtesy of Karen McKoy, MD.

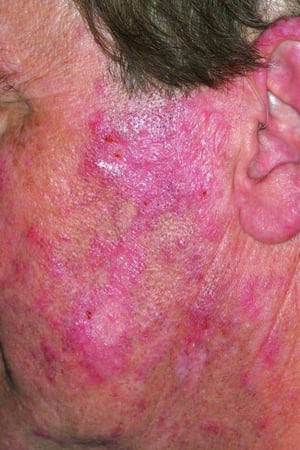
This photo shows chronic discoid lupus erythematosus with characteristic hyperkeratotic and erythematous plaques.
This photo shows chronic discoid lupus erythematosus with characteristic hyperkeratotic and erythematous plaques.
© Springer Science+Business Media
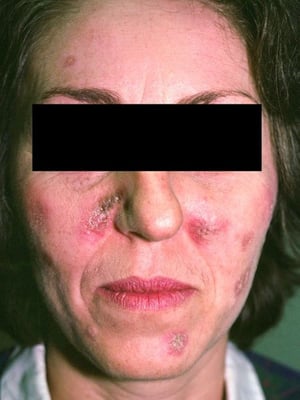
This photo shows erythematous scaling plaques and atrophic scars on the face resulting from discoid lupus erythematosus.
This photo shows erythematous scaling plaques and atrophic scars on the face resulting from discoid lupus erythematosus
Photo courtesy of Karen McKoy, MD.
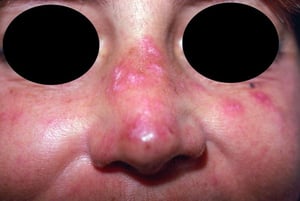
This photo shows erythematous scaling plaques on the face resulting from discoid lupus erythematosus.
This photo shows erythematous scaling plaques on the face resulting from discoid lupus erythematosus.
Photo courtesy of Karen McKoy, MD.


This photo shows erythematous and hyperpigmented scaling plaques with some central atrophic scarring on the back.
This photo shows erythematous and hyperpigmented scaling plaques with some central atrophic scarring on the back.
Photo courtesy of Karen McKoy, MD.


This photo shows multiple annular, hyperpigmented, scaling, and faintly erythematous plaques of the face and lips.
This photo shows multiple annular, hyperpigmented, scaling, and faintly erythematous plaques of the face and lips.
Photo courtesy of Karen McKoy, MD.


This photo shows multiple annular, hyperpigmented, scaling plaques and central depressed scarring.
This photo shows multiple annular, hyperpigmented, scaling plaques and central depressed scarring.
Photo courtesy of Karen McKoy, MD.


This photo shows multiple hyperpigmented plaques, some with slight follicular prominence and scale, on the face and neck. Involvement of the ear is common.
This photo shows multiple hyperpigmented plaques, some with slight follicular prominence and scale, on the face and nec
Photo courtesy of Karen McKoy, MD.
This photo shows discoid erythematous plaques causing atrophic scarring and resultant alopecia on the scalp.
This photo shows discoid erythematous plaques causing atrophic scarring and resultant alopecia on the scalp.
Photo courtesy of Karen McKoy, MD.
This photo shows chronic discoid lupus erythematosus with characteristic hyperkeratotic and erythematous plaques.
This photo shows chronic discoid lupus erythematosus with characteristic hyperkeratotic and erythematous plaques.
© Springer Science+Business Media
This photo shows erythematous scaling plaques and atrophic scars on the face resulting from discoid lupus erythematosus.
This photo shows erythematous scaling plaques and atrophic scars on the face resulting from discoid lupus erythematosus
Photo courtesy of Karen McKoy, MD.
This photo shows erythematous scaling plaques on the face resulting from discoid lupus erythematosus.
This photo shows erythematous scaling plaques on the face resulting from discoid lupus erythematosus.
Photo courtesy of Karen McKoy, MD.
This photo shows erythematous and hyperpigmented scaling plaques with some central atrophic scarring on the back.
This photo shows erythematous and hyperpigmented scaling plaques with some central atrophic scarring on the back.
Photo courtesy of Karen McKoy, MD.
This photo shows multiple annular, hyperpigmented, scaling, and faintly erythematous plaques of the face and lips.
This photo shows multiple annular, hyperpigmented, scaling, and faintly erythematous plaques of the face and lips.
Photo courtesy of Karen McKoy, MD.
This photo shows multiple annular, hyperpigmented, scaling plaques and central depressed scarring.
This photo shows multiple annular, hyperpigmented, scaling plaques and central depressed scarring.
Photo courtesy of Karen McKoy, MD.
This photo shows multiple hyperpigmented plaques, some with slight follicular prominence and scale, on the face and neck. Involvement of the ear is common.
This photo shows multiple hyperpigmented plaques, some with slight follicular prominence and scale, on the face and nec
Photo courtesy of Karen McKoy, MD.
Patients presenting with typical discoid lesions should be evaluated for SLE. Antibodies against dsDNA are almost invariably absent in DLE. Although it does not differentiate DLE from SLE, biopsy can rule out other disorders (eg, lymphoma, sarcoidosis). Biopsy should be done from the margin of an active skin lesion.
Early treatment of DLE can prevent permanent atrophy. Exposure to sunlight or ultraviolet light should be minimized (eg, using potent sunscreens when outdoors).
For limited disease, topical corticosteroid ointments (particularly for dry skin) or creams (less greasy than ointments) 3 to 4 times a day (eg, triamcinolone acetonide 0.1 or 0.5%, fluocinolone 0.025 or 0.2%, flurandrenolide 0.05%, betamethasone valerate 0.1%, and, particularly betamethasone dipropionate 0.05%) usually cause involution of small lesions; they should not be used excessively or on the face (where they cause skin atrophy). Resistant lesions can be covered with plastic tape coated with flurandrenolide. Alternatively, intradermal injection with triamcinolone acetonide 0.1% suspension (For limited disease, topical corticosteroid ointments (particularly for dry skin) or creams (less greasy than ointments) 3 to 4 times a day (eg, triamcinolone acetonide 0.1 or 0.5%, fluocinolone 0.025 or 0.2%, flurandrenolide 0.05%, betamethasone valerate 0.1%, and, particularly betamethasone dipropionate 0.05%) usually cause involution of small lesions; they should not be used excessively or on the face (where they cause skin atrophy). Resistant lesions can be covered with plastic tape coated with flurandrenolide. Alternatively, intradermal injection with triamcinolone acetonide 0.1% suspension (< 0.1 mL per site) may resolve lesions, but secondary atrophy frequently follows. A topical calcineurin inhibitor (eg, tacrolimus) may be used for areas such as the face or for lesions that have been resistant to topical corticosteroids. 0.1 mL per site) may resolve lesions, but secondary atrophy frequently follows. A topical calcineurin inhibitor (eg, tacrolimus) may be used for areas such as the face or for lesions that have been resistant to topical corticosteroids.
For more extensive disease or lesions that do not respond to topical or intralesional corticosteroids, antimalarials (eg, oral hydroxychloroquine 5 mg/kg once a day) are recommended, including for facial lesions.For more extensive disease or lesions that do not respond to topical or intralesional corticosteroids, antimalarials (eg, oral hydroxychloroquine 5 mg/kg once a day) are recommended, including for facial lesions.
If initial approaches fail, combination therapy with hydroxychloroquine 200 mg/day plus oral quinacrine 50 to 100 mg once a day or If initial approaches fail, combination therapy with hydroxychloroquine 200 mg/day plus oral quinacrine 50 to 100 mg once a day orhydroxychloroquine plus dapsone, methotrexate, mycophenolate mofetil, or azathioprine is used.plus dapsone, methotrexate, mycophenolate mofetil, or azathioprine is used.
Subacute cutaneous lupus erythematosus (SCLE)
Patients with SCLE develop extensive recurring rashes. Annular or papulosquamous lesions (psoriasiform lesions) may develop on the face, arms, and trunk. Lesions are usually photosensitive and can develop hypopigmentation but rarely scar. SCLE can be drug-induced, for example, triggered by antihypertensives (eg, diuretics, calcium channel blockers, beta blockers), proton pump inhibitors (eg, omeprazole, pantoprazole), anti-tumor necrosis factor (TNF) biologics (eg, adalimumab), and antifungals (eg, terbinafine).Patients with SCLE develop extensive recurring rashes. Annular or papulosquamous lesions (psoriasiform lesions) may develop on the face, arms, and trunk. Lesions are usually photosensitive and can develop hypopigmentation but rarely scar. SCLE can be drug-induced, for example, triggered by antihypertensives (eg, diuretics, calcium channel blockers, beta blockers), proton pump inhibitors (eg, omeprazole, pantoprazole), anti-tumor necrosis factor (TNF) biologics (eg, adalimumab), and antifungals (eg, terbinafine).
Patients may be antinuclear antibody (ANA)-positive or ANA-negative. Patients usually have antibodies to Ro (SSA). Infants whose mother has Ro antibodies may have congenital SCLE or congenital heart block.
A significant minority of patients with SCLE may develop SLE, with one study reporting almost 10% of patients (1). Thus, patients with SCLE should be monitored periodically for systemic involvement.
SCLE is treated similarly to DLE.


This photo shows a diffuse, erythematous rash on sun-exposed areas.

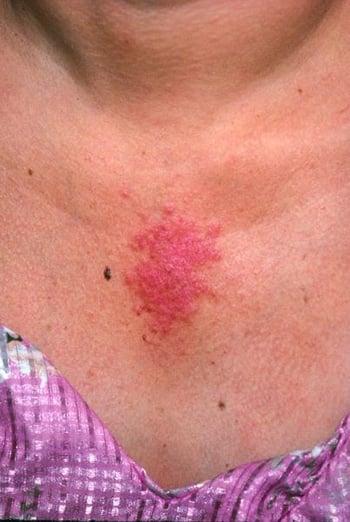
This photo shows papulosquamous lesions typical of subacute cutaneous lupus erythematosus.
SCLE reference
1. Alniemi DT, Gutierrez A Jr, Drage LA, Wetter DA. Subacute Cutaneous Lupus Erythematosus: Clinical Characteristics, Disease Associations, Treatments, and Outcomes in a Series of 90 Patients at Mayo Clinic, 1996-2011. Mayo Clin Proc. 2017;92(3):406-414. doi:10.1016/j.mayocp.2016.10.030
Drugs Mentioned In This Article





