

Microscopic polyangiitis is a systemic necrotizing vasculitis without immune globulin deposition (pauci-immune) that affects mainly small vessels. It may begin as a pulmonary-renal syndrome with rapidly progressing glomerulonephritis and alveolar hemorrhage, but the pattern of disease depends on the organs affected. Diagnosis is made by clinical findings and sometimes confirmed by biopsy. Treatment, which depends on disease severity, includes corticosteroids with other immunosuppressants.
(See also Overview of Vasculitis.)
Microscopic polyangiitis (MPA) is rare (approximately 6 cases/million) (1). Pathogenesis is unknown. Rarely, MPA can occur in association with hepatitis B.
MPA affects small vessels (including capillaries and postcapillary venules) and is pauci-immune (ie, immune globulin deposition is not seen on tissue biopsy), similar to granulomatosis with polyangiitis (GPA) and eosinophilic granulomatosis with polyangiitis (EGPA), which differentiates it from immune complex-mediated small-vessel vasculitides (eg, immunoglobulin A–associated vasculitis—formerly known as Henoch-Schönlein purpura) and cutaneous vasculitis.
Clinical manifestations of MPA can resemble those of granulomatosis with polyangiitis, and include alveolar hemorrhage, glomerulonephritis, and multiple mononeuropathy. However, granulomatous destructive lesions (eg, pulmonary cavitary lesions) are typically absent in MPA and the upper respiratory tract is usually affected minimally or not at all. In both disorders, antineutrophil cytoplasmic antibodies (ANCA) are frequently present.
General reference
1. Watts RA, Mooney J, Skinner J, Scott DG, Macgregor AJ. The contrasting epidemiology of granulomatosis with polyangiitis (Wegener's) and microscopic polyangiitis. Rheumatology (Oxford) 51(5):926-931, 2012. doi:10.1093/rheumatology/ker454
Symptoms and Signs of Microscopic Polyangiitis
Usually, a prodromal illness with systemic symptoms of fever, weight loss, myalgia, and arthralgia occurs. Other symptoms depend on which organs and systems are affected:
Renal: The kidneys are affected in up to 90% of patients. Hematuria, proteinuria (sometimes > 3 g/24 hours), and red blood cell casts are present. Without prompt diagnosis and treatment, renal failure may follow rapidly.
Cutaneous: Approximately one-third of patients have a purpuric rash at the time of the diagnosis. Nail bed infarcts and splinter hemorrhages may occur; digital ischemia occurs rarely.
Respiratory: If the lungs are affected, alveolar hemorrhage may occur and may be followed by pulmonary fibrosis. Rapid-onset dyspnea and anemia, with or without hemoptysis and bilateral patchy infiltrates (seen on chest radiograph), may be due to alveolar hemorrhage, a medical emergency that requires immediate treatment. Some patients with MPA may initially present with interstitial lung disease. Mild symptoms of rhinitis, epistaxis, and sinusitis may occur; however, if the upper respiratory tract is severely affected, the cause is more likely to be granulomatosis with polyangiitis.
Gastrointestinal (GI): GI symptoms include abdominal pain, nausea, vomiting, diarrhea, and bloody stools.
Neurologic: The peripheral nervous system is frequently affected, with multiple mononeuropathy (mononeuritis multiplex) that affects peripheral or cranial nerves. Rarely, cerebral hemorrhage, infarction, seizures, or headache results from cerebral vasculitis.
Cardiac: Rarely, the heart is affected.
Ocular: Eyes can be affected, usually with episcleritis.
Diagnosis of Microscopic Polyangiitis
Clinical findings
Tests for antineutrophil cytoplasmic antibodies and routine laboratory tests
Biopsy
Microscopic polyangiitis should be suspected in patients who have unexplained combinations of fever, weight loss, arthralgias, abdominal pain, alveolar hemorrhage, new-onset nephritic syndrome, new-onset multiple mononeuropathy, or new-onset polyneuropathy. Laboratory tests and sometimes imaging are done, but the diagnosis is usually confirmed by biopsy.
Tests include complete blood count, erythrocyte sedimentation rate (ESR), C-reactive protein, urinalysis, serum creatinine, and tests for antineutrophil cytoplasmic antibodies (ANCA). ESR, C-reactive protein levels, and white blood cell and platelet counts are elevated, reflecting systemic inflammation. Anemia of chronic disease is common. An acute drop in hematocrit suggests alveolar hemorrhage or hemorrhage in the gastrointestinal tract. Urinalysis with urinary sediment (to check for hematuria, proteinuria, and cellular casts) should be done, and serum creatinine should be measured periodically to check for renal involvement.
Immunofluorescence staining can detect ANCA; this test is followed by an enzyme-linked immunosorbent assay (ELISA) to check for specific antibodies. At least 70% of patients are ANCA-positive, usually perinuclear ANCA (p-ANCA) with antibodies against myeloperoxidase (1).
Biopsy of the most accessible involved tissue should be done to confirm vasculitis. Renal biopsy may detect focal segmental pauci-immune necrotizing glomerulonephritis with fibrinoid necrosis of the glomerular capillary wall, leading to formation of cellular crescents.
In patients with respiratory symptoms, chest imaging is done to check for infiltrates. Bilateral patchy infiltrates suggest alveolar hemorrhage even in patients without hemoptysis. CT is much more sensitive than radiograph.

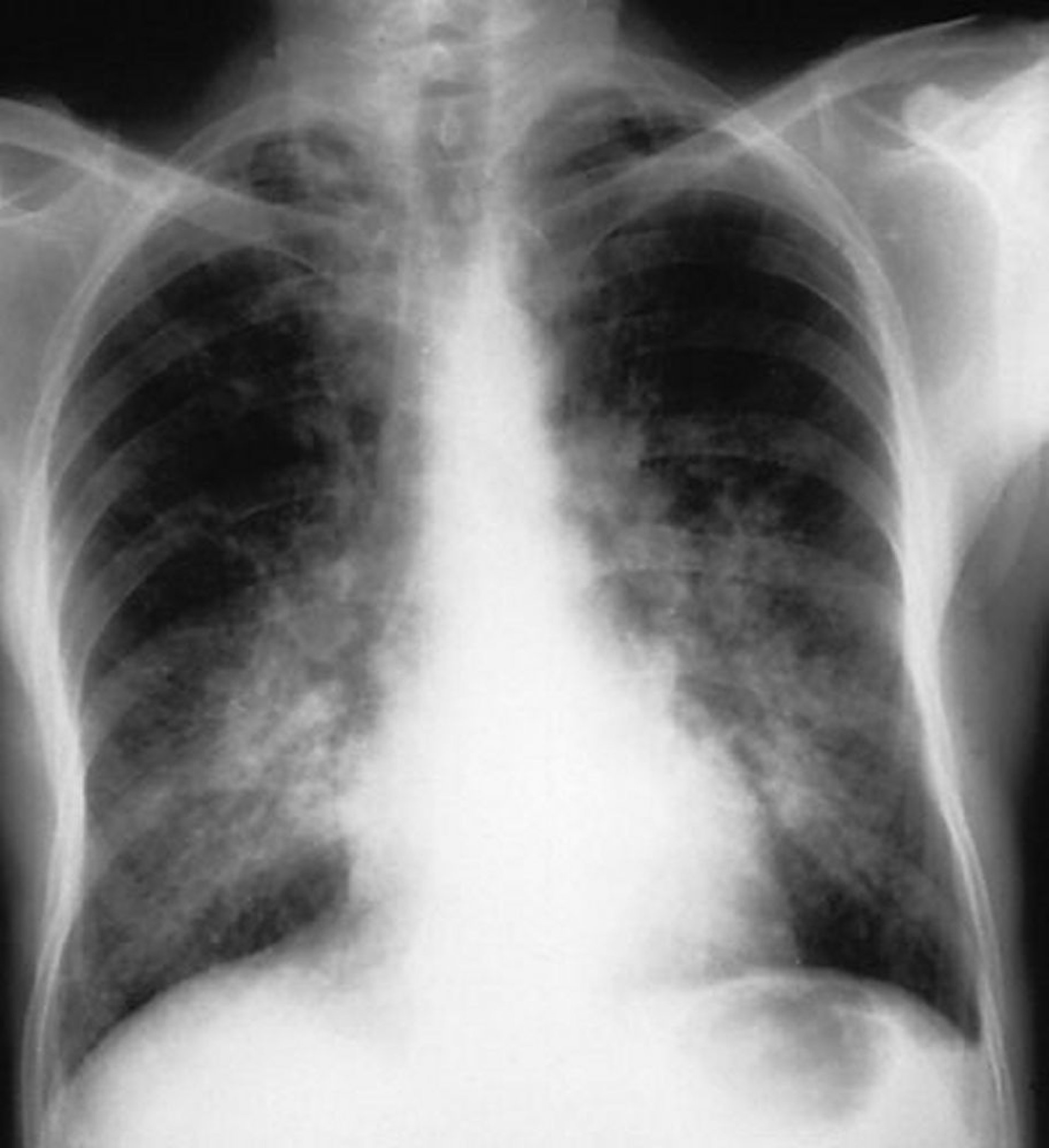
By permission of the publisher. From Cohen A, Glassock R. In Atlas of Diseases of the Kidney: Glomerulonephritis and Vasculitis. Edited by R Schrier (series editor), RJ Glassock, and AH Cohen. Philadelphia, Current Medicine, 1999.
If patients have dyspnea and bilateral infiltrates, bronchoscopy should be done immediately to check for alveolar hemorrhages and to exclude infection. Blood coming from both lungs and all bronchi, with more blood coming as the bronchoscope goes deeper in the airways, indicates active alveolar hemorrhage. Hemosiderin-laden macrophages appear within 24 to 72 hours after onset of hemorrhage and may persist for up to 2 months.
Diagnosis reference
1. Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42(3):421-430. doi:10.1002/1529-0131(199904)42:3<421::AID-ANR5>3.0.CO;2-6
Treatment of Microscopic Polyangiitis
When vital organs are affected, high-dose corticosteroids plus rituximab or cyclophosphamideWhen vital organs are affected, high-dose corticosteroids plus rituximab or cyclophosphamide
For less severe cases, corticosteroids plus rituximab or methotrexateFor less severe cases, corticosteroids plus rituximab or methotrexate
Treatment is the same as that for granulomatosis with polyangiitis, using high-dose corticosteroids in combination with another immunosuppressant.
Key Points
Microscopic polyangiitis is a rare small-vessel vasculitis.
Manifestations are variable and may include alveolar hemorrhage, multiple mononeuropathy, and glomerulonephritis.
Confirm the diagnosis by testing for antineutrophil cytoplasmic antibodies and by biopsy.
Treat with high-dose corticosteroids plus another immunosuppressant (eg, rituximab or cyclophosphamide for severe disease).Treat with high-dose corticosteroids plus another immunosuppressant (eg, rituximab or cyclophosphamide for severe disease).
Drugs Mentioned In This Article





