



Tricuspid atresia is absence of the tricuspid valve accompanied by a hypoplastic right ventricle. Associated anomalies are common and include atrial septal defect, ventricular septal defect, patent ductus arteriosus, pulmonic valve stenosis, and transposition of the great arteries. Presenting signs include cyanosis or signs of heart failure. The first heart sound (S1)is single and may be accentuated. The 2nd heart sound (S2) is usually single. Most infants have a murmur, the nature of which depends on the presence of associated anomalies. Diagnosis is by echocardiography. Cardiac catheterization may be needed. Definitive treatment is surgical repair.
(See also Overview of Congenital Cardiovascular Anomalies.)
Tricuspid atresia accounts for 1 to 3% of congenital heart anomalies.
The most common type (sometimes referred to as classic tricuspid atresia) includes a ventricular septal defect (VSD) and pulmonic stenosis, which results in decreased pulmonary blood flow, elevated right atrial pressure, and an obligatory right-to-left shunt at the atrial level through a stretched patent foramen ovale or an atrial septal defect (ASD), causing cyanosis (see figure ). Infrequently, classic tricuspid atresia involves a large VSD and mild pulmonic stenosis, resulting in pulmonary overcirculation.
In 12 to 25% of cases, the great arteries are transposed with a VSD and a normal pulmonic valve, with unrestricted pulmonary blood flow coming directly from the left ventricle, typically resulting in heart failure and pulmonary hypertension.
Thus, pulmonary blood flow may be increased or decreased with different forms of tricuspid atresia.
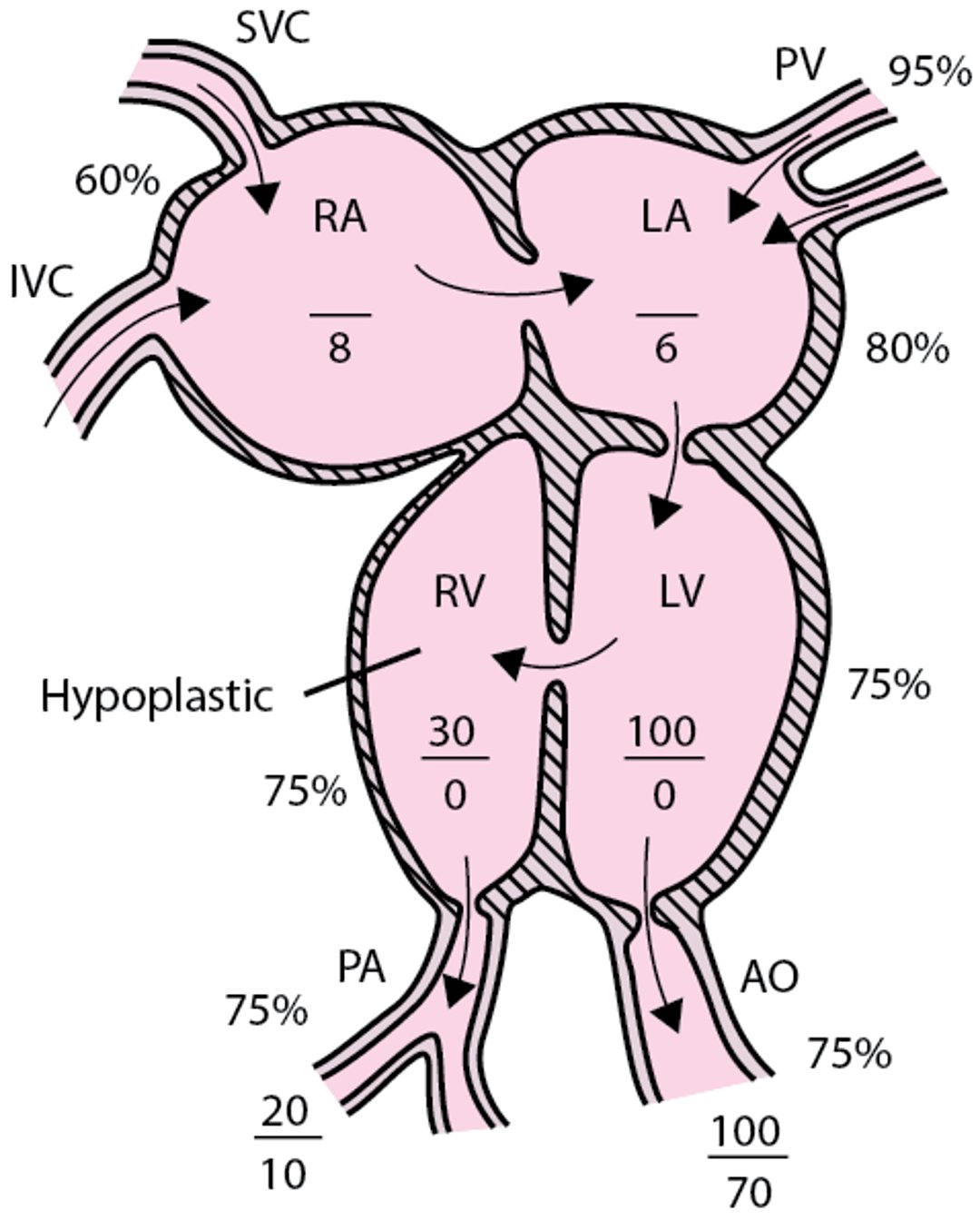
Tricuspid atresia with normally related great vessels
The tricuspid valve is absent, and the right ventricle is hypoplastic. An atrial communication must be present. AO = aorta; IVC = inferior vena cava; LA = left atrium; LV = left ventricle; PA = pulmonary artery; PV = pulmonary veins; RA = right atrium; RV = right ventricle; SVC = superior vena cava. |

Symptoms and Signs of Tricuspid Atresia
Infants with decreased pulmonary blood flow usually have mild-to-moderate cyanosis at birth, which increases, sometimes dramatically, over the first several months of life. Infants with increased pulmonary blood flow usually show signs of heart failure (eg, tachypnea, dyspnea with feeding, poor weight gain, diaphoresis) by age 4 to 6 weeks.


This photo shows clubbing of a patient's finger in comparison to a normal matched finger.
© Springer Science+Business Media
Physical examination usually detects a single prominent first heart sound (S1), a single second heart sound (S2) in patients with marked pulmonic valve stenosis or transposed great vessels, and a grade 2 to 3/6 holosystolic or early systolic murmur of a VSD at the lower left sternal border (see table ). A systolic ejection murmur of pulmonic stenosis or a continuous murmur of patent ductus arteriosus may be present at the upper left sternal border. A systolic thrill is rarely palpable. An apical diastolic rumble may be audible if pulmonary blood flow is markedly increased. Cyanosis, when present for > 6 months, may result in finger clubbing.
Diagnosis of Tricuspid Atresia
Chest x-ray and ECG
Echocardiography
Usually cardiac catheterization
Diagnosis of tricuspid atresia is suspected clinically, supported by chest x-ray and ECG, and established by 2-dimensional echocardiography with color flow and Doppler studies.
In the most common form, chest x-ray shows normal or slightly increased heart size, right atrial enlargement, and decreased pulmonary vascular markings. Occasionally, the cardiac silhouette resembles that of tetralogy of Fallot (with a boot-shaped heart and concave pulmonary artery segment). Pulmonary vascular markings may be increased and cardiomegaly may be present in infants with associated transposition of the great arteries. ECG characteristically shows left axis deviation (between 0° and −90°) and left ventricular hypertrophy. Left axis deviation is not usually present if there is associated transposition of the great arteries. Right atrial or combined atrial enlargement is also common.
Cardiac catheterization may be useful (particularly in older children) before the first palliative procedure to define hemodynamics and pulmonary artery anatomy.
Treatment of Tricuspid Atresia
For severely cyanotic neonates, prostaglandin E1 infusion
Rarely balloon atrial septostomy
Staged surgical repair
Most neonates with tricuspid atresia, although cyanotic, are well compensated in the first several weeks of life. In severely cyanotic neonates, prostaglandin E1 (beginning at 0.05 to 0.1 mcg/kg/minute IV) is infused to prevent closure of the ductus arteriosus or to reopen the constricted ductus before cardiac catheterization or surgical repair.
Rarely, when the patent foramen ovale/atrial septal defect is restrictive, balloon atrial septostomy (Rashkind procedure) may be done as part of the initial catheterization to decompress the right atrium and facilitate unrestricted right-to-left atrial shunting. In the balloon atrial septostomy, a balloon-tipped catheter is advanced into the left atrium through the patent foramen ovale. The balloon is inflated and abruptly withdrawn to the right atrium thereby enlarging the opening in the atrial septum. The procedure immediately improves systemic arterial oxygen saturation.
Some infants with transposition of the great arteries and signs of heart failure require medical treatment (eg, diuretics, digoxin, angiotensin-converting enzyme inhibitors).
Definitive repair requires staged operations. If intervention is needed for hypoxemia within the first 4 to 8 weeks of life, a modified Blalock-Taussig-Thomas shunt (connection of a systemic and a pulmonary artery by a synthetic tube) is done.
In infants with excess pulmonary blood flow and signs of heart failure, a pulmonary artery band may be placed to limit pulmonary blood flow. Otherwise, if the infant remains stable with good growth, the first procedure would be a bidirectional Glenn or a hemi-Fontan procedure (anastomosis between the superior vena cava and right pulmonary artery) at 3 to 6 months of age. A modified Fontan procedure is then done, usually between 1 year and 2 years.
The Fontan procedure involves diverting the inferior vena cava flow directly to the pulmonary artery using either a baffle created within the right atrium (lateral tunnel) or an extracardiac conduit that completely bypasses the right atrium. The proximal pulmonary root is ligated, which prevents anterograde flow across the pulmonary outflow tract, and an adequate interatrial opening is created, if not already present, to allow equalization of right and left atrial pressures and free communication between these chambers. A fenestration (small opening) is frequently made between the Fontan pathway and the right atrium. Right-to-left shunting from the Fontan pathway to the atria and left ventricle allows decompression of the systemic venous pressure and improvement in cardiac output, albeit at the expense of mild arterial desaturation. This approach has increased early survival rates to > 90%, 5-year survival rates to > 80%, and 10-year survival rates to >70%.
Endocarditis prophylaxis is recommended preoperatively and for at least 6 months after each surgical intervention and for as long as the patient remains cyanotic or has a residual defect adjacent to a surgical patch or prosthetic material.
Key Points
The tricuspid valve is absent, and the right ventricle is hypoplastic; these defects are fatal unless there is an opening between the atria along with a ventricular septal defect and/or patent ductus arteriosus.
Infants with decreased pulmonary blood flow have progressively worsening cyanosis; infants with increased pulmonary blood flow usually have heart failure (eg, tachypnea, dyspnea with feeding, poor weight gain, diaphoresis).
Relieve severe cyanosis during the newborn period by giving prostaglandin E1 infusion to keep the ductus arteriosus open.
Definitive treatment requires staged operations.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
American Heart Association: Common Heart Defects: Provides overview of common congenital heart defects for parents and caregivers
American Heart Association: Infective Endocarditis: Provides an overview of infective endocarditis, including summarizing prophylactic antibiotic use, for patients and caregivers
Drug Information for the Topic





