





Overproduction of growth hormone causes excessive growth. In children, the condition is called gigantism. In adults, it is called acromegaly.
Excessive growth hormone is almost always caused by a noncancerous (benign) pituitary tumor.
Children develop great stature, and adults develop deformed bones but do not grow taller.
Heart failure, weakness, and vision problems are common.
The diagnosis is based on blood tests and imaging of the skull and hands.
Computed tomography (CT) or magnetic resonance imaging (MRI) of the head is done to look for the cause.
A combination of surgery, radiation therapy, and medication therapy is used to treat the overproduction of growth hormone.
(See also Overview of the Pituitary Gland.)

Growth hormone is produced by the anterior lobe of the pituitary gland. Growth hormone stimulates the growth of bones, muscles, and many internal organs. Excessive growth hormone, therefore, leads to abnormally robust growth of all of these tissues. Overproduction of growth hormone is almost always caused by a noncancerous (benign) pituitary tumor (adenoma). Certain rare tumors of the pancreas and lungs also can produce hormones that stimulate the pituitary to produce excessive amounts of growth hormone, with similar consequences.
Symptoms of Gigantism and Acromegaly
If excessive production of growth hormone starts in childhood before the growth plates of the bones (the area at the ends of bones from which bone growth occurs) have closed, the condition causes gigantism. The long bones grow enormously. A person grows to unusually great stature, and the arms and legs lengthen. Puberty may be delayed, and the genitals may not develop fully.
In most cases, excessive production of growth hormone begins between the ages of 30 and 50 years, long after the growth plates of the bones have closed. Increased growth hormone in adults thus cannot increase the length of bones, but it does cause acromegaly, in which the bones become deformed rather than elongated. Because changes occur slowly, they are usually not recognized for years.
Did You Know?
|

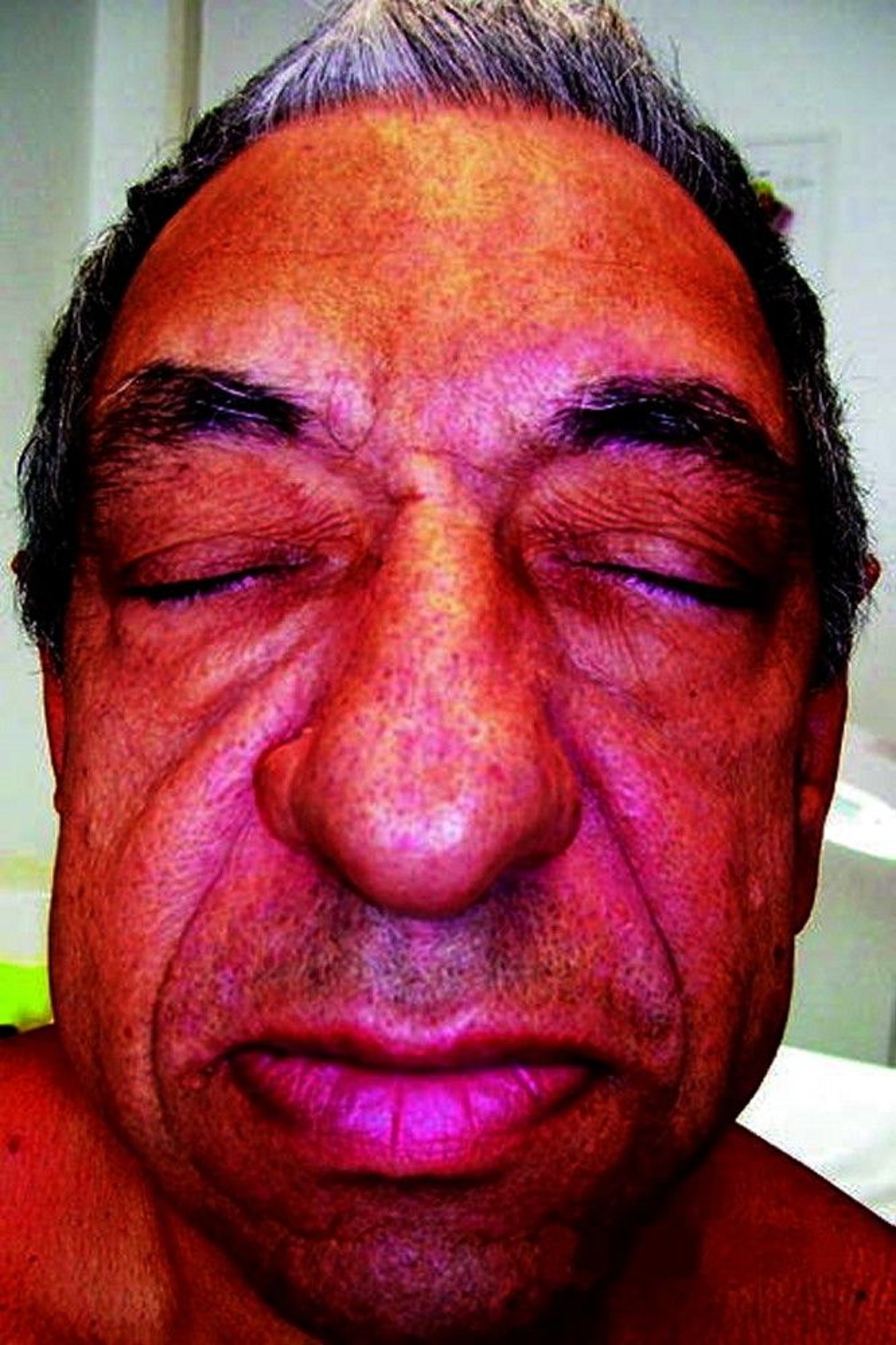
This photo shows a person who has an enlarged forehead and nose, protruding jaw, and thickened skin.
© Springer Science+Business Media
The person's facial features become coarse, and the hands and feet swell. Larger rings, gloves, shoes, and hats are needed. Overgrowth of the jawbone (mandible) can cause the jaw to protrude (prognathism). Cartilage in the voice box (larynx) may thicken, making the voice deep and husky. The ribs may thicken, creating a barrel chest. Joint pain is common. After many years, crippling degenerative arthritis may occur.


The woman on the right was diagnosed with gigantism as an infant. She is several feet taller than her mother on the left.
BETTINA CIRONE/SCIENCE PHOTO LIBRARY
In both gigantism and acromegaly, the tongue may enlarge and become more furrowed. Coarse body hair, which typically darkens, increases as the skin thickens. The sebaceous and sweat glands in the skin enlarge, producing excessive perspiration and often an offensive body odor.
The heart usually enlarges, and its function may be so severely impaired that heart failure occurs. Other organs in the body may be enlarged.
Sometimes a person feels disturbing sensations and weakness in the arms and legs as enlarging tissues compress the nerves. Nerves that carry messages from the eyes to the brain may also be compressed, causing loss of vision, particularly in the outer visual fields. The pressure on the brain may also cause severe headaches.
Nearly all women with acromegaly have irregular menstrual cycles. Some women produce breast milk even though they are not breastfeeding (galactorrhea) because of a related increase in the hormone prolactin. About one-third of men who have acromegaly develop erectile dysfunction.
There is also an increased likelihood of developing diabetes mellitus, high blood pressure (hypertension), sleep apnea, and certain tumors, particularly affecting the large intestine, which may become cancerous. Life expectancy is reduced in people with untreated acromegaly.
Diagnosis of Gigantism and Acromegaly
Blood tests
Imaging tests
In children, rapid growth may not seem abnormal at first. Eventually, however, the abnormality of the extreme growth becomes clear.
In adults, because the changes induced by high levels of growth hormone occur slowly, acromegaly often is not diagnosed until many years after the first symptoms appear. Serial photographs (those taken over many years) may help a doctor establish the diagnosis.
Imaging of the skull may show thickening of the bones and enlargement of the nasal sinuses. X-rays of the hands show thickening of the bones under the fingertips and swelling of the tissue around the bones.
Blood sugar levels and blood pressure may be high.
Computed tomography (CT) or magnetic resonance imaging (MRI) is usually done to look for abnormal growths in the pituitary gland. Because acromegaly is usually present for some years before being diagnosed, a tumor is seen on these scans in most people.

The diagnosis is confirmed by blood tests, which usually show high levels of both growth hormone and insulin-like growth factor 1 (IGF-1). Because growth hormone is released in short bursts and the levels of growth hormone often fluctuate dramatically even in people without acromegaly, a single high level of growth hormone in the blood is insufficient to make the diagnosis. Doctors must give something that would normally suppress growth hormone levels, most commonly a glucose drink, and show that normal suppression does not occur. This test is not necessary when the clinical features of acromegaly are obvious, the IGF-1 level is high, or a tumor is seen in the pituitary on CT or MRI.
Once acromegaly is diagnosed, additional testing is usually done to detect complications, such as diabetes, high blood pressure, or digestive system tumors, that may have developed.
Treatment of Gigantism and Acromegaly
Surgery
Radiation therapy
Medication
Stopping or reducing the overproduction of growth hormone is not easy. Doctors may need to use a combination of surgery, radiation therapy, and medication.
Surgery
Surgery to remove the pituitary tumor by an experienced surgeon is currently regarded as the best first treatment for most people with gigantism and acromegaly caused by a tumor. It results in an immediate reduction in tumor size and growth hormone production, most often without causing deficiency of other pituitary hormones.
Unfortunately, tumors are often large by the time they are found, and surgery alone does not usually cure the disorder. Long-term treatment with medication is often needed after surgery.
Radiation therapy is sometimes used as a follow-up treatment, particularly if a substantial amount of the tumor remains after surgery and acromegaly symptoms persist despite other treatments.
Medications
Medications can also be used to treat elevated growth hormone levels. Medications that act on the tumor to suppress growth hormone secretion and medications that block growth hormone from interacting with its receptor are available. Both types of medication reduce symptoms related to excessive growth hormone secretion.
The choice of medication is individualized to the person being treated, but most people receive treatment with a class of medications called somatostatin receptor ligands, which reduce growth hormone secretion, thereby reducing IGF-1 production. In many cases, these medications also shrink the tumor. These medications include octreotide, lanreotide, and pasireotide. Each is usually given once a month.
Pegvisomant, a growth hormone receptor blocker, is also used to treat acromegaly. It may be useful for people who do not respond to somatostatin receptor ligands. It lowers IGF-1 production by interfering with the action of the growth hormone coming from the tumor. It is given as a daily injection under the skin and is effective in most people when the dose is increased to adequate levels.
Occasionally, cabergoline is used to treat acromegaly, especially in milder cases when the serum IGF-1 level is not very elevated above normal. This medication is not as effective as somatostatin receptor ligands, but because it is taken by mouth rather than injection, it may be preferred by some people.
All of these medications are effective in controlling acromegaly in many people as long as they continue to be taken; however, they do not provide a cure.
Radiation therapy
Radiation therapy involves the delivery of single or multiple smaller doses of radiation to the tumor, which is less traumatic than surgery. However, it may take several years for radiation to have its full effect on reducing growth hormone secretion. People take somatostatin receptor ligands while they are waiting for radiation therapy to have its full effect.
Later on, radiation therapy often results in deficiencies of other pituitary hormones because normal tissue is often also affected.
Two types of radiation therapy may be used:
Conventional radiation therapy
Stereotactic radiation therapy
In conventional radiation therapy, small doses of radiation are delivered to the tissue several times over a period of 6 to 8 weeks. However, conventional radiation therapy may also damage healthy tissue that is near the tumor.
Stereotactic radiation therapy, in which the radiation comes from several different directions (so it does not always go through the same normal tissue), has been developed for use with multiple types of radiation used including linear accelerators, proton beam, and gamma knife.
Drug Information for the Topic





