



The long QT interval syndromes (LQTS) result from any congenital or acquired disorder of cardiac ion channel function or regulation (channelopathy) that prolongs ventricular myocyte action potential duration as reflected by prolongation of the rate-corrected QT interval on the ECG. Patients are at risk for torsades de pointes polymorphic ventricular tachycardia, which may cease spontaneously or degenerate into ventricular fibrillation. Diagnosis is by clinical criteria and ECG, sometimes with exercise and/or provocative testing. Treatment is avoidance of triggers, beta-blockade, and sometimes an implantable cardioverter-defibrillator and/or left cardiac sympathetic denervation.
Long QT interval syndromes can be acquired, congenital, or both. (For discussion of acquired causes, see Torsades de Pointes Ventricular Tachycardia). The incidence of congenital LQTS is at least 1 in 2000 (1, 2) and is the cause of approximately 15 to 20% of cardiac arrests that are unexplained after clinical evaluation that includes and ECG, an echocardiogram, and excluding coronary artery disease (3, 4). It is the most common of the cardiac channelopathies. (See also Overview of Arrhythmias.)
General references
1. Schwartz PJ, Crotti L. Long QT Syndrome. N Engl J Med. 2025;393(20):2023-2034. doi:10.1056/NEJMra2400853
2. Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761-1767. doi:10.1161/CIRCULATIONAHA.109.863209
3. Krahn AD, Healey JS, Chauhan V, et al. Systematic assessment of patients with unexplained cardiac arrest: Cardiac Arrest Survivors With Preserved Ejection Fraction Registry (CASPER). Circulation. 2009;120(4):278-285. doi: 10.1161/CIRCULATIONAHA.109.853143
4. Rucinski C, Winbo A, Marcondes L, et al. A Population-Based Registry of Patients With Inherited Cardiac Conditions and Resuscitated Cardiac Arrest. J Am Coll Cardiol. 2020;75(21):2698-2707. doi:10.1016/j.jacc.2020.04.004
Pathophysiology of Long QT Interval Syndromes
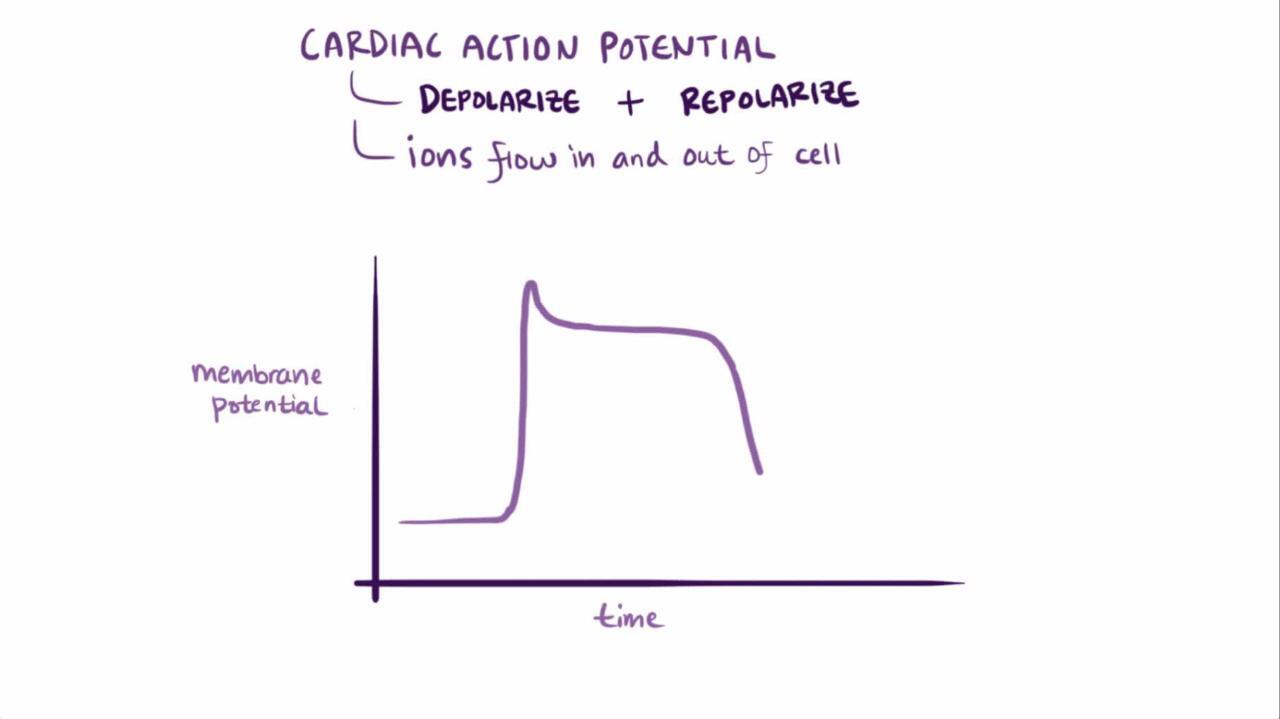
The congenital long QT interval syndromes result from genetic disorders of cardiac ion channel function or regulation (channelopathies) that prolong ventricular myocyte action potential duration as reflected by prolongation of the rate-corrected QT interval on the ECG (QTc, typically calculated using Bazett formula). More than 15 types have been described.

The dysfunction may involve:
Loss of function of repolarizing potassium current channels OR
Gain of function of depolarizing sodium or depolarizing calcium current channels
Prolongation of action potentials increases the probability of transmembrane voltage oscillations occurring during the depolarized myocyte action potential (early afterdepolarizations). If the action potential durations of myocytes in a local area vary, these oscillations may reactivate neighboring myocytes that have repolarized and thus create torsades de pointes ventricular tachycardia (TdP VT). The risk of TdP VT is dependent on the degree of QTc prolongation, particularly if it is > 0.50 second (1, 2).
LQTS type 3 (see Etiology) also causes early-onset paroxysmal atrial fibrillation, and its occurrence is associated with an increased risk of presumed arrhythmic syncope, cardiac arrest, and sudden cardiac death.
Predisposing factors for arrhythmia
The occurrence of TdP VT is favored by any condition that further prolongs action potential duration (ie, the QT interval), including female sex, bradycardia, hypokalemia, hypomagnesemia, and hypothyroidism. Other risk factors include slow or irregular ventricular rate, acute intracranial events (eg, bleeding, stroke, traumatic brain injury), eating disorders, organophosphate poisoning, and structural heart disease (especially acute ischemia, myocarditis, and ventricular hypertrophy). Many medications are risk factors, particularly class Ia, Ic, and III antiarrhythmic medications, as well as other medications, including tricyclic antidepressants, phenothiazines, and certain antivirals and antifungals (see CredibleMeds for up-to-date information). Often, several of these factors are present (3).
Pathophysiology references
1. Drew BJ, Ackerman MJ, Funk M, et al. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation. 2010;121(8):1047-1060. doi:10.1161/CIRCULATIONAHA.109.192704
2. Follansbee CW, Landstrom AP, Aziz PF. Cardiac Channelopathies in the Pediatric Patient: Long QT Syndrome. Card Electrophysiol Clin. 2025;17(4):631-644. doi:10.1016/j.ccep.2025.07.011
3. Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259(1):59-69. doi: 10.1111/j.1365-2796.2005.01589.x
Etiology of Long QT Interval Syndromes
Long QT interval syndromes are classified based on the specific gene that has mutated. However, a specific genetic abnormality is identified in only 70 to 85% of cases (1); likelihood of detecting an abnormality varies depending on clinical factors present.
More than 15 forms of LQTS have been described; the vast majority of cases are LQTS1, LQTS2, or LQTS3. These 3 forms are inherited as autosomal dominant disorders with incomplete penetrance:
Long QT syndrome type 1 (LQTS1): Loss-of-function mutation of gene KCNQ1, which encodes an adrenergic-sensitive Kv7.1 channel responsible for the slow outward potassium current (IKs) (40 to 55% of gene-positive cases) (2)
Long QT syndrome type 2 (LQTS2): Loss-of-function mutation of gene KCNH2, which encodes the hERG channel responsible for the rapid outward potassium current (IKr) (30 to 45% of gene-positive cases (2)
Long QT syndrome type 3 (LQTS3): Gain-of-function mutation of gene SCN5A, which encodes the Nav1.5 channel responsible for the inward sodium current (INa) (5 to 10% of gene-positive cases) (2)
Rare forms of LQTS with additional clinical features have been described including Jervell and Lange Nielsen syndrome (with congenital sensorineural deafness), Andersen-Tawil syndrome (with period paralysis and craniofacial dysmorphisms), and Timothy syndrome (with craniofacial dysmorphisms, immunodeficiency, congenital heart disease, developmental delay, and syndactyly).

Etiology references
1. Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. J Arrhythm. 2022;38(4):491-553. doi: 10.1002/joa3.12717
2. Schwartz PJ, Crotti L. Long QT Syndrome. N Engl J Med. 2025;393(20):2023-2034. doi:10.1056/NEJMra2400853
Symptoms and Signs of Long QT Interval Syndromes
The LQTS are asymptomatic unless TdP VT occurs, which can cause palpitations, near-syncope, syncope, or sudden death. Some patients experience myoclonic jerks during syncope; they may incorrectly have a diagnosis of epilepsy pursued. Because the ventricular action potential duration decreases with increasing heart rate, TdP VT is often self-terminating. However, it may degenerate into ventricular fibrillation and cause cardiac arrest.
Some forms of LQTS are more associated with certain triggers than others:
LQTS1: Physical stress/exertion, particularly swimming, or emotional stress
LQTS2: Sudden loud noises like an alarm clock
LQTS3: Resting or sleep
In patients with LQTS, there is a 5-year risk of a first episode of life-threatening arrhythmia ranging from approximately 0.3% to 17% (1). Overall lifetime risk of a cardiac event varies from < 30% to ≥ 80% (2). Predictors include the following:
Age (higher in children than in adults)
Sex (higher in post-pubertal females than in pre-pubertal females)
Use of beta-blocker therapy (higher in patients not taking beta-blockers)
Syncope (higher in those with a history of presumed arrhythmic syncope)
QTc interval (especially if ≥ 0.50 second)
Genotype (if available; higher in LQTS2 and LQTS3 than in LQTS1)
An online risk calculator is available for patients who have not yet had a life-threatening arrhythmia (University of Rochester Long QT Syndrome Risk Calculator) (1). A simplified risk calculator based only on the QTc interval and genotype (1-2-3 Long QT Syndrome Risk Calculator) is also available (3, 4).
Symptoms and signs references
1. Wang M, Peterson DR, Pagan E, et al. Assessment of absolute risk of life-threatening cardiac events in long QT syndrome patients. Front Cardiovasc Med. 2022;9:988951. doi:10.3389/fcvm.2022.988951
2. Giudicessi JR, Ackerman MJ. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr Probl Cardiol. 2013;38(10):417-455. doi:10.1016/j.cpcardiol.2013.08.001
3. Mazzanti A, Maragna R, Vacanti G, et al. Interplay Between Genetic Substrate, QTc Duration, and Arrhythmia Risk in Patients With Long QT Syndrome. J Am Coll Cardiol. 2018;71(15):1663-1671. doi:10.1016/j.jacc.2018.01.078
4. Mazzanti A, Trancuccio A, Kukavica D, et al. Independent validation and clinical implications of the risk prediction model for long QT syndrome (1-2-3-LQTS-Risk). Europace. 2022;24(4):614-619. doi:10.1093/europace/euab238
Diagnosis of Long QT Interval Syndromes
Characteristic clinical and electrocardiographic manifestations
Sometimes exercise testing
Sometimes ambulatory ECG monitoring
Rarely provocative testing using IV epinephrine or isoproterenol
Genetic testing
Screening of relatives
Diagnosis should be considered in patients with unexplained cardiac arrest or syncope or a family history of such when the affected people do not have structural heart disease. Occasionally, documentation of TdP VT during a symptomatic episode (or incidentally) leads to the diagnosis. LQTS should also be considered in people who are discovered to have a long QT interval when ECG is performed for other reasons. Fetal or neonatal bradycardia or 2:1 atrioventricular block may also be the initial presentation of a congenital long QT syndrome. Finally, the diagnosis may be made by genetic testing performed as part of familial screening of relatives of patients with congenital long QT syndrome.
A long QT interval is diagnosed by ECG showing prolongation of the rate-corrected QT interval (QTc). In adults, normal QTc values are < 0.43 second for males and < 0.45 second for females and are considered prolonged when > 0.44 to 0.45 second for males or > 0.46 to 0.47 second for females (1, 2). Proper measurement technique, including validation of computer-generated measurements, is important and can affect the result significantly (2, 3). Similarly, correction for rate can be challenging at particularly low, high, or variable heart rates; multiple rate correction techniques exist. Although Bazett formula has limitations (particularly overestimation at faster heart rates), it remains commonly used.
Torsades de Pointes Ventricular Tachycardia

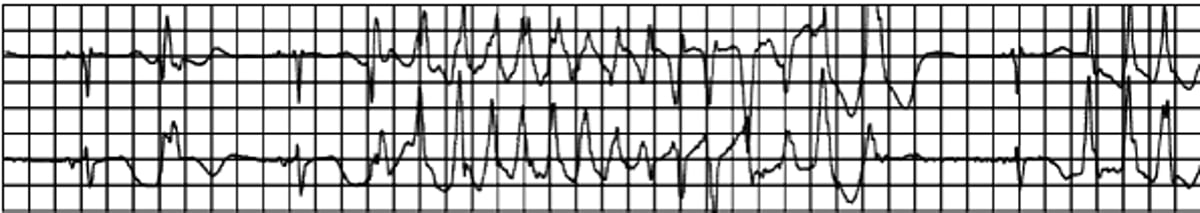
Some forms of LQTS are also associated with particular ECG patterns:
LQTS1: Wide T waves
LQTS2: Low voltage, notched T-waves
LQTS3: Long-ST segment with normal appearing T-waves
Neither arrhythmia triggers nor ECG findings are very specific and should not be used to confirm the type or to direct genetic testing.
Given the multiplicity of factors affecting the QTc, a normal QTc, found in 25 to 50% of gene mutation-positive LQTS cases, does not exclude the diagnosis (3, 4, 5). Nevertheless, at the moment of torsade de pointes VT, the QTc is essentially always prolonged.
When a patient has a resting QTc interval ≥ 0.46 second and documented torsade de pointes VT or syncope/cardiac arrest with stress in the absence of other causes of a prolonged QT interval, the diagnosis of a congenital long QT interval syndrome is established (6). Patients with borderline QT intervals suspected of having LQTS, or those with a LQTS genotype but a normal resting QT interval, should have exercise testing, because some abnormalities appear only during or after exercise (5, 7). Ambulatory ECG monitoring may also disclose transient ventricular repolarization abnormalities. In patients with a normal QTc interval, provocative testing with IV isoproterenol or epinephrine may disclose a concealed long QTc, nevertheless, due to a high false-positive rate, poor reproducibility, and substantial variation in interpretation, provocative testing is not routinely recommended for diagnosis (6).
Because not all patients with a long QT interval have congenital long QT syndrome and because not all patients with a congenital long QT syndrome have a long QT interval on any given ECG, scoring systems based on the original Schwartz score have been developed and modified over time to establish a diagnosis of a congenital LQTS (see table ) (8). Probability is estimated as low (score ≤ 1), intermediate (score 1.5 to 3.0), or high (score ≥ 3.5) based on clinical, ECG, and exercise testing criteria, provided that the patient is not presently exposed to any other causes of QT-interval prolongation. The modified Schwartz score can also be used to establish candidacy for genetic testing. Patients with a low probability of a congenital LQTS do not need genetic testing, but patients in whom the probability is intermediate or high do. Patients with a high probability of a congenital LQTS without a detected genetic abnormality may be considered to represent one of the 15 to 30% of patients with an unidentified mutation. Intermediate-probability patients who are gene mutation–negative are followed closely with repeated electrocardiographic examinations including ECG, ambulatory cardiac monitoring, and exercise testing (8).
Modified Schwartz Score for Long QT Syndrome (LQTS)*
Criteria | Points |
|---|---|
Patient history | |
Syncope with stress† | 2 |
Syncope without stress† | 1 |
Congenital deafness | 0.5‡ |
Family history | |
Family member with known LQTS¶ | 1 |
Family member with unexplained cardiac death before age 30 years ¶ | 0.5 |
Electrocardiography§ | |
QTc ≥ 480 msec | 3‡ |
QTc 460-479 msec | 2 |
QTc 450-459 msec (males only) | 1 |
QTc ≥ 480 msec during 4th minute of recovery from an exercise test | 1 |
Torsades de pointes arrhythmia§ | 2 |
T wave alternans | 1 |
Notched T wave in 3 leads | 1 |
Resting heart rate < 2nd percentile for age | 0.5 |
* SCORE: ≤ 1 low probability; 1.5–3 intermediate probability; ≥ 3.5 high probability | |
† Mutually exclusive (ie, if syncope occurs with stress, no points are given for syncope without stress) | |
‡ In the modified long QT syndrome diagnostic score:
| |
¶ Mutually exclusive | |
§ If torsades de pointes present, do not score syncope | |
Data from Schwartz PJ, Crotti L: QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124:2181-2184. doi: 10.1161/CIRCULATIONAHA.111.062182; Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation. 2018;138(13):e272-e391. doi:10.1161/CIR.0000000000000549; and Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43(40):3997-4126. doi:10.1093/eurheartj/ehac262 | |
Even the currently used Modified Schwartz scoring approach does not include genotype, however. To further improve the sensitivity of LQTS diagnosis another modification was recently proposed (the Modified Long QT Syndrome Diagnostic Score) (6). This score establishes a lower diagnostic threshold (score ≥ 3.0) to diagnose LQTS, and sets the number of points allocated for both a resting QTc interval of ≥ 0.48 second on repeated ECGs and a pathogenic LQTS mutation to 3.5 points, so that either alone is sufficient for a LQTS diagnosis.
First-degree relatives of the index case should have clinical evaluation (ie, to detect symptoms suggestive of arrhythmia) and ECG. Thereafter, the first-degree relatives of any newly identified patients undergo similar assessment (cascade screening). Genetic testing is performed when the index case has a known mutation. If the index case is gene-mutation negative, first-degree relatives have exercise testing if the results could alter the Schwartz score probability class. Family members may undergo periodic electrocardiographic evaluation if their genetic status is unknown and the diagnosis remains unclear.
Diagnosis references
1. Goldenberg I, Moss AJ, Zareba W. QT interval: how to measure it and what is "normal". J Cardiovasc Electrophysiol. 2006;17(3):333-336. doi: 10.1111/j.1540-8167.2006.00408.x
2. Rautaharju PM, Surawicz B, Gettes LS, et al. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol. 2009;53(11):982-991. doi:10.1016/j.jacc.2008.12.014
3. Krahn AD, Laksman Z, Sy RW, et al. Congenital Long QT Syndrome. JACC Clin Electrophysiol. 2022;8(5):687-706. doi:10.1016/j.jacep.2022.02.017
4. Mazzanti A, Maragna R, Vacanti G, et al. Interplay Between Genetic Substrate, QTc Duration, and Arrhythmia Risk in Patients With Long QT Syndrome. J Am Coll Cardiol. 2018;71(15):1663-1671. doi:10.1016/j.jacc.2018.01.078
5. Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124(20):2187-2194. doi:10.1161/CIRCULATIONAHA.111.028258
6. Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43(40):3997-4126. doi:10.1093/eurheartj/ehac262
7. Horner JM, Horner MM, Ackerman MJ. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm. 2011;8(11):1698-1704. doi:10.1016/j.hrthm.2011.05.018
8. Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124(20):2181-2184. doi: 10.1161/CIRCULATIONAHA.111.062182
Treatment of Long QT Interval Syndromes
Treatment of any VT/VF
Alleviation of predisposing causes and triggers, especially electrolyte abnormalities and use of certain medications
Beta-blockade
Sometimes mexiletine
Sometimes an implantable cardioverter-defibrillator (ICD)
Sometimes left cardiac sympathetic denervation
Details of treatment of torsade de pointes ventricular tachycardia are discussed elsewhere but include defibrillation for torsade-induced VF and magnesium sulfate IV. Patients with frequent or long runs of torsades de pointes ventricular tachycardia may benefit from treatment to shorten the QT interval by increasing the heart rate using temporary pacing. Unlike medication-induced acquired torsade de pointes, the mainstay of acute IV pharmacotherapy is IV beta-blocker rather than IV isoproterenol.
Long-term treatment to prevent sudden death includes avoidance of genotype-specific triggers (including careful discussion of strenuous exercise in LQTS1 and LQTS2) and QTc-prolonging conditions or medications (see CredibleMeds for up-to-date information) (1). When possible, clinicians should stop any predisposing medications and prescribe alternatives. Patients who choose to participate in strenuous exercise or competitive sports should be counseled on the need for appropriate cautions (eg, availability of an automated external defibrillator during training and competition). Fever should be controlled to reduce associated sympathetic nervous system stimulation.
Beta-blocker therapy is recommended for patients who have LQTS with QTc interval prolongation or who are symptomatic (most effective with LQTS1 or LQTS2) (1,2). Beta-blocker therapy is reasonable in asymptomatic patients with LQTS and a normal QTc interval (1, 2). When a beta-blocker is used, a long-acting, non-selective beta-blocker (eg, nadolol, slow-release propranolol) is preferred. When beta-blocker therapy is ineffective at relieving symptoms in patients with LQTS3, mexiletine is added (2).
Permanent pacing to increase the basal ventricular rate and to prevent post-extrasystolic pauses may reduce the probability of recurrent TdP VT. An ICD is indicated in patients who have been resuscitated after cardiac arrest and in those who have cardiac syncope despite therapy with a beta-blocker. An ICD may also be considered for patients with a QTc > 500 msec while on a beta-blocker even when asymptomatic and for those with a high-risk profile based on the 1-2-3 LQTS Risk Calculator (1, 2). (see table ).
Left heart (stellate ganglion) denervation may also be used (3).
Treatment references
1. Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43(40):3997-4126. doi:10.1093/eurheartj/ehac262
2. Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation. 2018;138(13):e272-e391. doi:10.1161/CIR.0000000000000549
3. Savastano S, Schwartz PJ. Blocking nerves and saving lives: Left stellate ganglion block for electrical storms. Heart Rhythm. 2023;20(7):1039-1047. doi: 10.1016/j.hrthm.2022.11.025
Key Points
Congenital long QT interval syndromes may cause torsades de pointes ventricular tachycardia, ventricular fibrillation, and sudden death.
Numerous factors, particularly use of certain medications, increase the risk of ventricular arrhythmias.
Diagnosis is based on a combination of genetic testing and clinical and electrocardiographic criteria, including exercise and sometimes provocative testing.
Family members should be screened.
Long-term management includes avoidance of triggers, use of beta-blockers, and sometimes mexiletine, permanent pacing, an implantable cardioverter-defibrillator, or stellate ganglion denervation.
Drug Information for the Topic





