

Amyloidosis is any of a group of disparate conditions characterized by extracellular deposition of insoluble fibrils composed of misaggregated proteins. These proteins may accumulate locally, causing relatively few symptoms, or widely, involving multiple organs and causing severe multiorgan failure. Amyloidosis can occur de novo or be secondary to various infectious, inflammatory, or malignant conditions. Diagnosis is by biopsy of affected tissue; the amyloidogenic protein is typed using a variety of immunohistologic and biochemical techniques. Treatment varies with the type of amyloidosis.
Amyloid fibrils are made of normally soluble misfolded proteins that aggregate into oligomers and then insoluble fibrils. A number of normal (wild-type) and mutant proteins are susceptible to such misfolding and aggregation (amyloidogenic proteins), thus accounting for the wide variety of causes and types of amyloidosis.
Amyloid deposits are composed of small (approximately 10 nm diameter), insoluble fibrils that form congophilic beta-pleated sheets that can be identified by x-ray diffraction. In addition to the fibrillar amyloid protein, the deposits also contain serum amyloid P component and glycosaminoglycans.
Amyloid deposits stain pink with hematoxylin and eosin, contain carbohydrate constituents that stain with periodic acid-Schiff dye or with Alcian blue, but most characteristically have apple-green birefringence under polarized light microscopy after Congo red staining. On autopsy inspection, affected organs may appear waxy.
For amyloidosis to develop, in addition to production of amyloidogenic proteins, there is probably also a failure of the normal clearance mechanisms for such misfolded proteins. The amyloid deposits themselves are metabolically inert but interfere physically with organ structure and function. However, some prefibrillar oligomers of amyloidogenic proteins have direct cellular toxicity, an important component of disease pathogenesis.
Etiology of Amyloidosis
In systemic amyloidosis, circulating amyloidogenic proteins form deposits in a variety of organs. Major systemic types include
AL amyloidosis: Caused by acquired overexpression of clonal immunoglobulin light chains
ATTR variant (ATTRv) amyloidosis (hereditary amyloidosis): Caused by inheritance of a mutant gene encoding a protein prone to misfolding, most commonly transthyretin (TTR)
ATTRwt (wild-type ATTR) amyloidosis: Caused by misfolding and aggregation of wild-type TTR
AA amyloidosis: Caused by aggregation of an acute phase reactant, serum amyloid A
Amyloidosis caused by aggregation of beta-2-microglobulin can occur in patients on long-term hemodialysis, but the incidence has declined with use of high-flow dialysis membranes. There is a rare hereditary form of beta-2-microglobulin amyloidosis due to a mutation to the relevant gene.
Localized forms of amyloidosis appear to be caused by local production and deposition of an amyloidogenic protein (most often immunoglobulin light chains) within the affected organ rather than by deposition of circulating proteins. Frequently involved sites include the central nervous system (eg, in Alzheimer disease), skin, upper or lower airways, lung parenchyma, bladder, eyes, and breasts.
AL amyloidosis
AL amyloidosis is caused by overproduction of an amyloidogenic immunoglobulin light chain in patients with a monoclonal plasma cell or other B cell lymphoproliferative disorder (1). Light chains can also form nonfibrillar tissue deposits (ie, light chain deposition disease). Rarely, immunoglobulin heavy chains form amyloid fibrils (called AH amyloidosis).
Common sites for amyloid deposition include the skin, nerves, heart, gastrointestinal tract (including the tongue), kidneys, liver, spleen, and blood vessels. Usually, a low-grade plasmacytosis is present in the bone marrow, which is similar to that in multiple myeloma, although most patients do not have true multiple myeloma (with lytic bone lesions, hypercalcemia, renal tubular casts, and anemia). However, approximately 10 to 20% of patients with multiple myeloma develop AL amyloidosis.
Hereditary amyloidosis
Hereditary amyloidosis is caused by inheritance of a gene encoding a mutated aggregation-prone serum protein, usually a protein abundantly produced by the liver.
Serum proteins that can cause hereditary amyloid include transthyretin (TTR), apolipoprotein A-I, apolipoprotein A-II, lysozyme, fibrinogen, gelsolin, and cystatin C. A form that is speculated to be familial is caused by the serum protein leukocyte chemotactic factor 2 (LECT2); however, a specific inherited gene mutation for this latter type has not been clearly demonstrated.
Amyloidosis caused by mutant TTR (ATTRv) is the most common type of hereditary amyloidosis. More than 130 mutations of the TTR gene have been associated with amyloidosis. The most prevalent mutation, V30M, is common in Portugal, Sweden, Brazil, and Japan, and a V122I mutation is present in approximately 4% of American and Caribbean Black people. Disease penetrance and age of onset are highly variable but are consistent within families and ethnic groups (2).
ATTRv causes peripheral sensorimotor neuropathy and autonomic neuropathy, chronic kidney disease, and cardiomyopathy. Carpal tunnel syndrome commonly precedes other neurologic disease manifestations. Vitreous deposits may develop due to production of mutant TTR by the retinal epithelium, or leptomeningeal deposits may develop as the choroid plexus produces mutant TTR. When cardiomyopathy is the predominant manifestation of TTR deposition in the heart, it is referred to as transthyretin amyloid cardiomyopathy (ATTR-CM).
ATTRwt amyloidosis
ATTRwt amyloidosis is caused by aggregation and deposition of wild-type TTR, principally targeting the heart.
ATTRwt is increasingly recognized as a cause of infiltrative cardiomyopathy in older men. Approximately 16% of patients with aortic stenosis undergoing transcatheter aortic valve replacement (3) and 13% of those hospitalized for heart failure with preserved ejection fraction (HFpEF) also have transthyretin amyloid cardiomyopathy, in this case designated as wATTR-CM to denote deposition of wild-type TTR in the heart (4). Soft-tissue manifestations of ATTRwt amyloidosis, including carpal tunnel syndrome, bicipital tendon rupture, rotator cuff tears, and spinal stenosis, may precede clinical expression of infiltrative cardiomyopathy by years.
The genetic and epigenetic factors leading to ATTRwt amyloidosis are unknown. Because ATTRwt and AL amyloidosis both can cause cardiomyopathy, and because amyloidogenic monoclonal gammopathies may be present in patients > 55 years of age, it is essential to accurately type the amyloid so that patients with ATTRwt amyloidosis are not inappropriately treated with chemotherapy (which is used for AL amyloidosis).
AA amyloidosis
This form can occur secondary to several infectious, inflammatory, and malignant conditions and is caused by aggregation of isoforms of the acute-phase reactant serum amyloid A.
Common causative infections include
Predisposing inflammatory conditions include
Inherited periodic fever syndromes such as familial Mediterranean fever
Castleman disease
Inflammatory cytokines (eg, interleukin [IL]-1, tumor necrosis factor [TNF], IL-6) that are produced in these disorders or ectopically by tumor cells cause increased hepatic synthesis of serum amyloid A.
AA amyloidosis shows a predilection for the kidneys, spleen, liver, adrenal glands, and lymph nodes. Involvement of the heart or peripheral and autonomic nerves occurs late in the disease course.
Localized amyloidosis
Localized amyloidosis outside the brain is most frequently caused by deposits of clonal immunoglobulin light chains; within the brain, amyloid beta protein predominates.
Localized amyloid deposits typically involve the airways and lung tissue, bladder and ureters, skin, breasts, and eyes. Rarely, other locally produced proteins cause amyloidosis, such as keratin isoforms that can form deposits locally in the skin. Clonal immunoglobulin light chains produced by mucosal-associated lymphoid tissue in the gastrointestinal tract, airways, and bladder can lead to localized AL in those organs.
Amyloid beta protein deposits in the brain contribute to Alzheimer disease or cerebrovascular amyloid angiopathy. Other proteins produced in the central nervous system can misfold, aggregate, and damage neurons, leading to neurodegenerative diseases (eg, Parkinson disease, Huntington disease).
Etiology references
1. Sanchorawala V. Systemic Light Chain Amyloidosis. N Engl J Med. 2024;390(24):2295-2307. doi:10.1056/NEJMra2304088
2. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
3. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
4. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al: Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 36(38):2585-2594, 2015. doi:10.1093/eurheartj/ehv338
Symptoms and Signs of Amyloidosis
Symptoms and signs of systemic amyloidosis are nonspecific, often resulting in delays in diagnosis. Suspicion of amyloidosis should be increased in patients with a progressive multisystem disease process.
Renal amyloid deposits typically occur in the glomerular membrane leading to proteinuria, but in approximately 15% of cases the tubules are affected, causing azotemia with minimal proteinuria. These processes can progress to nephrotic syndrome with marked hypoalbuminemia, edema, and anasarca or to end-stage kidney disease.
Hepatic involvement causes painless hepatomegaly, which may be massive. Liver tests typically suggest intrahepatic cholestasis with elevation of alkaline phosphatase and later bilirubin, although jaundice is rare. Occasionally, portal hypertension develops, with resulting esophageal varices and ascites.
Airway and laryngeal involvement leads to dyspnea, hoarseness, wheezing, hemoptysis, or airway obstruction.
Infiltration of the myocardium causes a restrictive cardiomyopathy, eventually leading to diastolic dysfunction and heart failure; heart block or arrhythmia may occur. Hypotension is common.
Peripheral neuropathy with paresthesias of the toes and fingers is a common presenting manifestation in AL and ATTRv amyloidoses. Autonomic neuropathy may cause orthostatic hypotension, erectile dysfunction, sweating abnormalities, urinary retention, and gastrointestinal motility disturbances.
Cerebrovascular amyloid angiopathy most often causes spontaneous cerebral hemorrhage, but some patients have brief, transient neurologic symptoms.
Gastrointestinal amyloid may cause motility abnormalities of the esophagus and small and large intestines. Gastric atony, malabsorption, bleeding, or pseudo-obstruction may also occur. Macroglossia is common in AL amyloidosis.
Soft tissue amyloid involvement characteristically precedes clinical expression of ATTRwt amyloid cardiomyopathy. Manifestations of soft tissue amyloid disease include carpal tunnel syndrome, trigger finger, bicipital tendon rupture, and spinal stenosis.

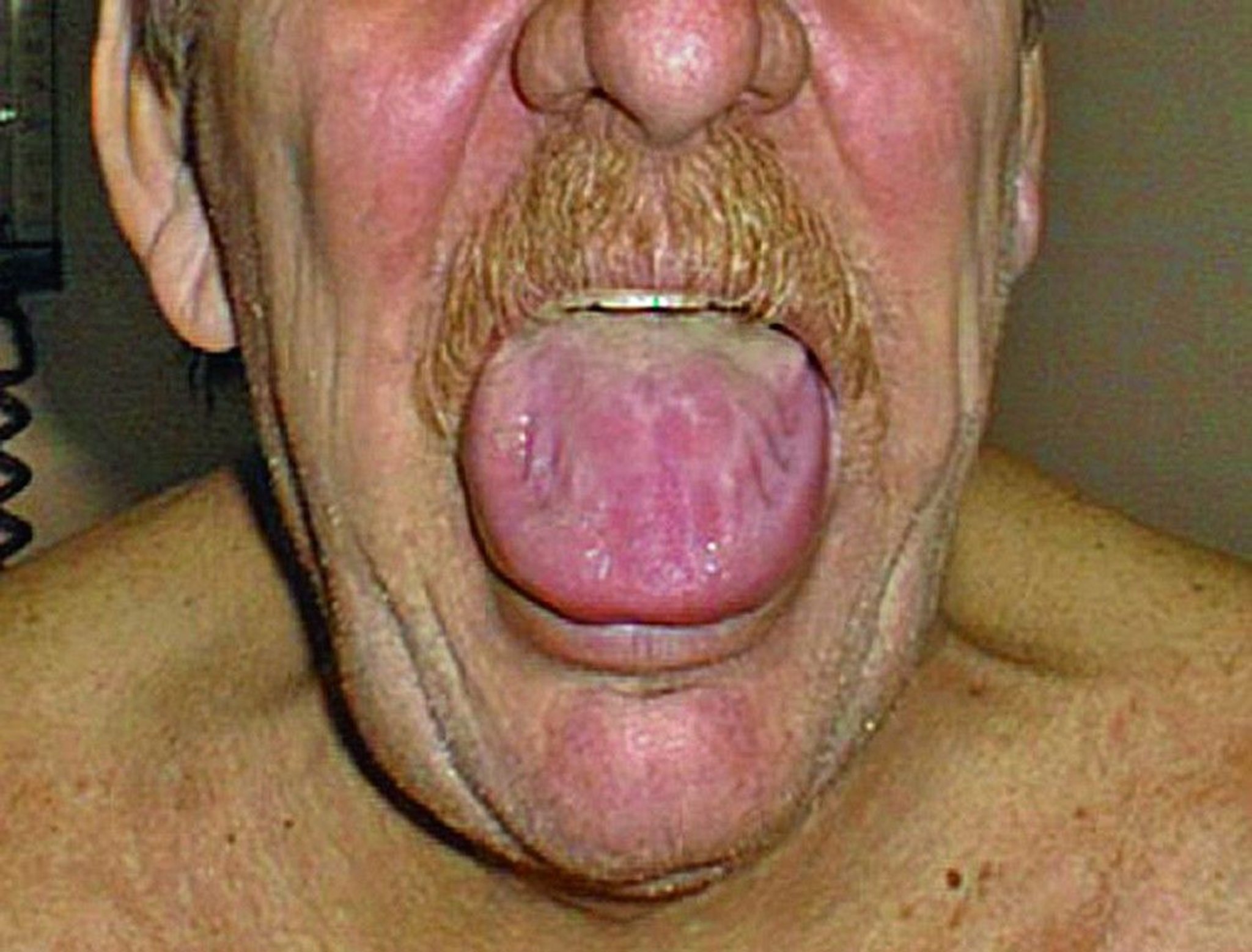
© Springer Science+Business Media
Amyloidosis of the thyroid gland may cause a firm, symmetric, nontender goiter resembling that found in Hashimoto thyroiditis. Other endocrinopathies can also occur.
Lung involvement (mostly in AL amyloidosis) can be characterized by focal pulmonary nodules and cysts, tracheobronchial lesions, pleural effusions, or diffuse alveolar-septal (interstitial) deposits.
Amyloid vitreous opacities and bilateral scalloped pupillary margins develop in several hereditary amyloidoses.
Other manifestations include bruising, often around the eyes (raccoon eyes), caused by amyloid deposits in blood vessels. Amyloid deposits cause weakening of the blood vessels, which may rupture after minor trauma, such as sneezing or coughing.
Diagnosis of Amyloidosis
Biopsy
Amyloid typing
Testing for organ involvement
Biopsy
Diagnosis of amyloidosis is made by demonstration of fibrillar deposits in an involved organ. Aspiration of subcutaneous abdominal fat detects amyloid deposits in approximately 80% of patients with AL but less than 25% of patients with ATTRwt (1). If the fat biopsy result is negative, a clinically involved organ should be biopsied. The diagnostic sensitivity of kidney and heart biopsies is nearly 100% when these organs are clinically involved. Tissue sections are stained with Congo red dye and examined with a polarizing microscope for characteristic birefringence. Nonbranching 10-nm fibrils can also be recognized by electron microscopy on biopsy specimens from the heart or kidneys.
Nuclear scanning using bone-avid tracers can diagnose ATTR amyloid cardiomyopathy without heart biopsy, provided AL amyloidosis is excluded.
Amyloid typing
After amyloidosis has been confirmed by biopsy, the type is determined using a variety of techniques. For some types of amyloidosis, immunohistochemistry or immunofluorescence may be diagnostic, but false-positive typing results occur. Other useful techniques include gene sequencing for hereditary amyloidosis and biochemical identification by mass spectrometry to accurately identify protein variants in amyloid deposits (the most sensitive and specific method).
If AL amyloidosis is suspected, patients should be evaluated for an underlying plasma cell disorder using quantitative measurement of serum free immunoglobulin light chains, qualitative detection of serum or urine monoclonal light chains using immunofixation electrophoresis (serum protein electrophoresis and urine protein electrophoresis are insensitive in patients with AL amyloidosis), and a bone marrow biopsy with flow cytometry or immunohistochemistry to establish plasma cell clonality.
Patients with > 10% clonal plasma cells should be tested to see if they meet criteria for multiple myeloma, including screening for lytic bone lesions, anemia, renal insufficiency, and hypercalcemia.
Organ involvement
Patients are screened for organ involvement beginning with noninvasive testing:
Kidneys: Urinalysis; measurement of serum blood urea nitrogen (BUN), creatinine, and albumin; estimated glomerular filtration rate (eGFR); and 24-hour urine collection for protein electrophoresis (UPEP)
Liver: Liver tests
Lungs: Chest radiograph, chest computed tomography (CT), and pulmonary function tests
Heart: Electrocardiogram (ECG) and measurement of serum biomarkers such as brain (B-type) natriuretic peptide (BNP) or N-terminal-pro-BNP (NT-proBNP) and troponin
Cardiac involvement can be suggested by low voltage on ECG (caused by a thickened ventricle) and/or dysrhythmias. If cardiac involvement is suspected because of symptoms, in addition to ECG findings and cardiac biomarkers, echocardiography is done to measure diastolic relaxation and global longitudinal strain (a measure of left ventricular systolic function) and to screen for biventricular hypertrophy. In ambiguous cases, cardiac magnetic resonance imaging (MRI) can be done to detect persistent subendocardial gadolinium enhancement, a characteristic finding. Cardiac technetium pyrophosphate nuclear scans improve detection of ATTR amyloid heart disease and can avoid the need for heart biopsies provided blood tests exclude AL amyloidosis (2, 3).
Diagnosis references
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Treatment of Amyloidosis
Supportive care
Type-specific treatment
There are specific treatments for most forms of amyloidosis, although some therapies are investigational. For all forms of systemic amyloidosis, supportive care measures can help relieve symptoms and improve quality of life.
Supportive care
Supportive care measures are directed at the affected organ system:
Renal: Patients with nephrotic syndrome and edema should be treated with salt and fluid restriction and loop diuretics; because of the ongoing protein loss, protein intake should not be restricted. Kidney transplantation is an option when the underlying disease process is controlled; it can provide long-term survival comparable to that in other renal diseases.
Cardiac: Patients with cardiomyopathy should be treated with salt and fluid restriction and loop diuretics. Other medications for heart failure, including digoxin, angiotensin-converting enzyme (ACE) inhibitors or , including digoxin, angiotensin-converting enzyme (ACE) inhibitors orangiotensin II receptor blockers (ARBs), calcium channel blockers, and beta-blockers, are poorly tolerated and contraindicated. Heart transplantation has been successful in carefully selected patients with AL and both types of ATTR amyloidosis and severe cardiac involvement. To prevent recurrence in the transplanted heart, patients with AL amyloidosis must be given aggressive chemotherapy directed at the clonal plasma cell disorder, and patients with symptomatic ATTR amyloid polyneuropathy or cardiomyopathy should be considered for anti-TTR therapies.
Gastrointestinal: Patients with diarrhea may benefit from loperamide. Those with early satiety and gastric retention may benefit from metoclopramide. Patients with diarrhea may benefit from loperamide. Those with early satiety and gastric retention may benefit from metoclopramide.
Nervous system: In patients with peripheral neuropathy, gabapentin, pregabalin, or duloxetine may relieve pain., gabapentin, pregabalin, or duloxetine may relieve pain.
Orthostatic hypotension often improves with high doses of midodrine; this medication can cause urinary retention in older males, but the medication complication of supine hypertension is rarely a problem in this population. Support stockings can also help, and fludrocortisone can be used in patients without peripheral edema, anasarca, or heart failure. In patients with refractory orthostatic hypotension, midodrine, fludrocortisone, or droxidopa may be added.often improves with high doses of midodrine; this medication can cause urinary retention in older males, but the medication complication of supine hypertension is rarely a problem in this population. Support stockings can also help, and fludrocortisone can be used in patients without peripheral edema, anasarca, or heart failure. In patients with refractory orthostatic hypotension, midodrine, fludrocortisone, or droxidopa may be added.
AL amyloidosis
For AL amyloidosis:
Prompt initiation of anti-plasma cell therapy is essential to preserve organ function and prolong life.
Most medications used for multiple myeloma have been used in AL amyloidosis; choice of medication, dose, and schedule often must be modified when organ function is impaired.
High-dose IV melphalan, combined with autologous High-dose IV melphalan, combined with autologousstem cell transplantation can be highly effective in selected patients (1).
For most patients, initial therapy typically includes daratumumab (an anti-CD38 monoclonal antibody) in combination with bortezomib (a proteasome inhibitor), dexamethasone, and an alkylating agent (eg, cyclophosphamide), referred to as Dara-CyBorD. This approach is supported by data from a randomized trial (ANDROMEDA) comparing the addition of For most patients, initial therapy typically includes daratumumab (an anti-CD38 monoclonal antibody) in combination with bortezomib (a proteasome inhibitor), dexamethasone, and an alkylating agent (eg, cyclophosphamide), referred to as Dara-CyBorD. This approach is supported by data from a randomized trial (ANDROMEDA) comparing the addition ofdaratumumab (Dara-CyBorD) to cyclophosphamide, bortezomib, and dexamethasone (CyBorD) in patients with newly diagnosed AL amyloidosis (excluding those with New York Heart Association [NYHA] class III and IV heart failure [see table NYHA Classification of Heart Failure], N-terminal pro-B-type natriuretic protein [NTproBNP] > 8,500 pg/mL [> 1005 pmol/L], and eGFR < 20 mL/minute/1.73 m2) (2). The addition of daratumumab resulted in an unprecedented higher rate of hematologic response. Hematologic response is based on monoclonal protein in serum and urine levels determined by immunofixation electrophoresis and serum light chain levels with kappa/lambda ratios. Longer-term follow up of patients in this clinical trial showed improvement in progression-free and overall survival and decreased major organ deterioration with Dara-CyBorD at a median follow up of 61 months.
Other regimens for patients with relapsing or refractory disease may include agents such as lenalidomide.Other regimens for patients with relapsing or refractory disease may include agents such as lenalidomide.
All available treatments target clonal B-cells or plasma cells in AL amyloidosis. Studies of antifibril antibodies, such as birtamimab and CAEL-101, are in progress (3).
Localized AL amyloidosis can be treated with low-dose external beam radiation therapy because plasma cells are highly radiosensitive.
ATTRv (hereditary) amyloidosis
For ATTR amyloidosis:
Liver transplantation
Tetramer-stabilizing medications
Gene silencing medications
Liver transplantation—which replaces the primary site of synthesis of the mutant protein with a new organ producing normal TTR—can be effective in certain TTR mutations if done at disease onset (early neuropathy and no heart involvement). Transplantation later in the course of the disease often leads to progressive amyloid cardiomyopathy and neuropathy due to the misfolding and deposition of wild-type TTR protein onto pre-existing amyloid deposits.
Several medications have been shown to stabilize TTR tetramers circulating in the plasma, inhibiting TTR misfolding and fibril formation and effectively slowing neurologic disease progression while preserving quality of life. These TTR stabilizers include diflunisal, a widely available generic anti-inflammatory drug, tafamidis (Several medications have been shown to stabilize TTR tetramers circulating in the plasma, inhibiting TTR misfolding and fibril formation and effectively slowing neurologic disease progression while preserving quality of life. These TTR stabilizers include diflunisal, a widely available generic anti-inflammatory drug, tafamidis (4, 5), and acoramidis (), and acoramidis (6). Tafamidis and acoramidis are used in the treatment of ATTR amyloid cardiomyopathy (7).
TTR gene silencing using anti-sense RNA or RNA interference to block translation of TTR mRNA efficiently reduces serum levels of TTR, improves neurologic outcomes in approximately 50% of patients, and, in some patients, appears capable of repairing injured nerves (8, 9). The gene silencing medications, patisiran, vutrisiran, and eplontersen, are available to treat patients with ATTR amyloid polyneuropathy. ). The gene silencing medications, patisiran, vutrisiran, and eplontersen, are available to treat patients with ATTR amyloid polyneuropathy.
Clinical trials of vutrisiran and eplontersen, second-generation gene silencers, demonstrated improved functional outcomes in patients with familial amyloid polyneuropathy (Clinical trials of vutrisiran and eplontersen, second-generation gene silencers, demonstrated improved functional outcomes in patients with familial amyloid polyneuropathy (9, 10). Data from 2 additional trials indicate that gene silencers also effectively treat cardiomyopathy in patients with ATTRv amyloidosis (11, 12).
ATTRwt amyloidosis
For ATTRwt amyloidosis:
Tetramer-stabilizing and gene silencing medications
TTR stabilization using tafamidis or acoramidis in patients with ATTRv or ATTRwt amyloid cardiomyopathy has been shown to decrease all-cause mortality and cardiovascular-related hospitalizations (TTR stabilization using tafamidis or acoramidis in patients with ATTRv or ATTRwt amyloid cardiomyopathy has been shown to decrease all-cause mortality and cardiovascular-related hospitalizations (5, 6). Both agents are used in the treatment of all ATTR amyloid cardiomyopathy. A clinical trial demonstrated that the TTR gene silencer vutrisiran effectively treats the cardiomyopathy that occurs in patients with ATTRwt amyloidosis as well as the cardiomyopathy that occurs in patients with ATTRv amyloidosis characterized by the mutant protein (). Both agents are used in the treatment of all ATTR amyloid cardiomyopathy. A clinical trial demonstrated that the TTR gene silencer vutrisiran effectively treats the cardiomyopathy that occurs in patients with ATTRwt amyloidosis as well as the cardiomyopathy that occurs in patients with ATTRv amyloidosis characterized by the mutant protein (10, 11, 12).
Unlike hereditary ATTR amyloidosis, liver transplantation is not effective for patients with ATTRwt because the amyloidogenic protein is a structurally normal TTR.
AA amyloidosis
For AA amyloidosis caused by familial Mediterranean fever, oral colchicine is effective. For AA amyloidosis caused by familial Mediterranean fever, oral colchicine is effective.
For other AA types, treatment is directed at the underlying infection, inflammatory disease, or cancer.
Colchicine or anti-IL1, anti-IL6, or anti-TNF medications may be used to interrupt the cytokine signaling, diminishing the inflammatory process driving hepatic production of serum amyloid A. Colchicine or anti-IL1, anti-IL6, or anti-TNF medications may be used to interrupt the cytokine signaling, diminishing the inflammatory process driving hepatic production of serum amyloid A.
Treatment references
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Gillmore JD, Judge DP, Cappelli F, et al: Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N Engl J Med 390(2):132–142, 2024. doi:10.1056/NEJMoa2305434
7. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
8. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
9. Coelho T, Marques Jr W, Dasgupta NR, et al: Eplontersen for hereditary transthyretin amyloidosis with polyneuropathy. JAMA 330:1448–1458, 2023. doi:10.1001/jama.2023.18688
10. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
11. Fontana M, Berk JL, Gillmore JD, et al: Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. N Engl J Med 392(1):33–44, 2025. doi:10.1056/NEJMoa2409134
12. Maurer MS, Kale P, Fontana M, et al: Patisiran treatment in patients with transthyretin cardiac amyloidosis. N Engl J Med 389(17):1553–1565, 2023. doi:10.1056/NEJMoa2300757
Prognosis for Amyloidosis
Prognosis depends on the type of amyloidosis and the organ system involved, but with appropriate disease-specific and supportive care, many patients have an excellent life expectancy.
AL amyloidosis complicated by severe cardiomyopathy has the poorest prognosis, with median survival of < 1 year. Untreated ATTR amyloidosis usually progresses to end-stage cardiac or neurologic disease within 5 to 15 years. ATTRwt was once thought to have the slowest progression of any systemic amyloidosis involving the heart; however, patients with ATTRwt do progress to symptomatic heart failure and death within a median of 4 years from biopsy diagnosis (1).
Prognosis in AA amyloidosis depends largely upon the effectiveness of treatment of the underlying infectious, inflammatory, or malignant disorder.
Prognosis reference
1. Connors LH, Sam F, Skinner M, et al. Heart Failure Resulting From Age-Related Cardiac Amyloid Disease Associated With Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation 2016;133(3):282-290. doi:10.1161/CIRCULATIONAHA.115.018852
Key Points
Amyloidosis is a group of disorders in which certain misfolded proteins aggregate into insoluble fibrils that are deposited within organs, causing dysfunction.
Many different proteins are prone to misfold; some of these proteins are produced by a genetic defect or by certain disease states, whereas others involve immunoglobulin light chains produced by monoclonal plasma cell or other B-cell lymphoproliferative disorders.
The amyloidogenic protein determines the amyloid type and clinical course of disease, although the clinical manifestations of the different types may overlap.
Many organs can be affected, but cardiac involvement carries a particularly poor prognosis; amyloid cardiomyopathy typically leads to diastolic dysfunction, heart failure, and heart block or arrhythmia.
Diagnosis is by biopsy; type of amyloidosis is determined by a variety of immunologic, genetic, and biochemical tests. Mass spectrometry is the most sensitive and specific method for amyloid typing.
Appropriate supportive care helps relieve symptoms and improves quality of life; organ transplantation can help selected patients.
Treat the underlying process; for AL amyloidosis due to plasma cell or lymphoproliferative disorders, chemotherapy can be highly effective; for AA amyloidosis, anti-infectious and anti-inflammatory medications can help.
For hereditary ATTRv amyloidosis, small molecule stabilizer therapeutics and gene-silencing medications inhibit or potentially reverse neurologic deterioration; for patients with amyloid cardiomyopathy (ATTRv or ATTRwt), small molecule stabilizer medications have been shown to decrease all-cause mortality and cardiovascular-related hospitalizations.
Drugs Mentioned In This Article


