



Bronchiectasis refers to dilation, thickening, and destruction of larger bronchi caused by chronic infection and inflammation. Common causes are cystic fibrosis, immune defects, and recurrent infections, though some cases seem to be idiopathic. Common symptoms are chronic cough and purulent sputum expectoration with or without dyspnea. Symptoms may worsen and may include fever during acute exacerbations. Diagnosis is based on history and imaging, usually involving high-resolution computed tomography, though standard chest x-radiographs may be diagnostic. Treatment and prevention of acute exacerbations are with bronchodilators, clearance of secretions, antibiotics, and management of complications, such as hemoptysis and further lung damage due to resistant or opportunistic infections. Treatment of underlying disorders is important whenever possible.
Etiology of Bronchiectasis
Bronchiectasis is estimated to affect 680/100,000 individuals annually worldwide (1). Bronchiectasis is best considered the common end-point of various disorders that cause chronic airway inflammation. Bronchiectasis may be diffuse (affecting many areas of the lungs) or focal (appearing in only 1 or 2 lung areas).
Bronchiectasis develops most often in patients with genetic, immunologic, or anatomic defects that affect the airways. The cause of many cases appears initially to be idiopathic, probably partly because onset is so slow that the triggering problem is not readily evident at the time bronchiectasis is recognized. However, after thorough investigation using genetic and immunologic testing, an etiology is more often found in these idiopathic cases (2).
Cystic fibrosis (CF) is commonly associated with diffuse bronchiectasis that classically predominates in the upper lobes, and previously undiagnosed CF may account for up to 20% of idiopathic cases. Even patients who are heterozygous, who typically have no clinical manifestations of CF, may have an increased risk of bronchiectasis.
Bronchiectasis also often coexists with more common conditions like chronic obstructive pulmonary disease (COPD) and asthma. It is an increasingly recognized complication of chronic, recurrent aspiration and gastroesophageal reflux.
Immunodeficiencies such as common variable immunodeficiency (CVID) may also lead to diffuse disease.
In regions where tuberculosis is common, bronchiectasis is a common complication, particularly in patients with impaired immune function due to undernutrition or human immunodeficiency virus (HIV) infection.
Rare abnormalities in airway structure may lead to diffuse bronchiectasis.
Congenital defects in mucociliary clearance such as primary ciliary dyskinesia (PCD) syndromes may also be a cause, explaining almost 3% of cases that were previously thought to be idiopathic.
Bronchiectasis sometimes complicates autoimmune disorders, such as rheumatoid arthritis, systemic lupus erythematosus (SLE), Sjögren syndrome, and inflammatory bowel disease. It can also occur in patients with hematologic malignancy or organ transplant, or it may be due to the immunocompromise associated with treatment in these conditions.
Allergic bronchopulmonary aspergillosis (ABPA), a hypersensitivity reaction to Aspergillus species that occurs most commonly in people with asthma, but sometimes in patients with CF, can cause or contribute to bronchiectasis.
Focal or localized bronchiectasis can develop as a result of untreated pneumonia or obstruction in the larger airways (eg, due to foreign bodies, tumors, postsurgical changes, lymphadenopathy). Mycobacteria (tuberculous or nontuberculous) can cause focal bronchiectasis and colonize the lungs of patients with bronchiectasis due to other disorders (see table ).
Factors Predisposing to Bronchiectasis
Category | Examples and Comments |
|---|---|
Infections | |
Bacterial | Haemophilus influenzae Pseudomonas aeruginosa Moraxella catarrhalis Staphylococcus aureus Streptococcus pneumoniae Mycoplasma pneumoniae Bordetella pertussis Klebsiella species |
Fungal | Aspergillus species (especially A. fumigatus) Histoplasma capsulatum |
Mycobacterial | Mycobacterium tuberculosis Nontuberculous mycobacteria |
Viral | Adenovirus Influenza Measles Respiratory syncytial virus SARS-CoV-2 Herpes simplex virus |
Airway obstruction | |
Cancer | Endobronchial lesion |
Extrinsic compression | Due to tumor mass or lymphadenopathy |
Foreign body | Aspirated or intrinsic (eg, broncholith) |
Mucoid impaction | Allergic bronchopulmonary aspergillosis |
Postoperative | After lobar resection, due to kinking or twisting of remaining lobes |
Congenital disorders | |
If severe, can cause bronchiectasis | |
Ciliary defects | Can cause bronchiectasis, sinusitis, otitis media, and male infertility Kartagener syndrome (clinical triad of dextrocardia, sinus disease, situs inversus) |
Causes viscous secretions due to defects in sodium and chloride transport Often complicated by P. aeruginosa or S. aureus colonization | |
Autosomal dominant connective tissue disorder | |
Immunodeficiencies | |
Primary | Chronic granulomatous disease Complement deficiencies Hypogammaglobulinemia, particularly common variable immunodeficiency (CVID) |
Secondary | HIV infection Hematologic malignancy Immunosuppressants Undernutrition |
Systemic rheumatic disorders | |
Commonly causes bronchiectasis (frequently subclinical), more often in men and in patients with long-standing rheumatoid arthritis | |
Bronchiectasis possibly due to increased viscosity of bronchial mucus, which leads to obstruction, poor clearance, and chronic infection | |
Bronchiectasis in up to 20% of patients via unclear mechanisms | |
Bronchopulmonary complications occurring after onset of inflammatory bowel disease in up to 85% and before onset in 10 to 15% Bronchiectasis more common in ulcerative colitis but can occur in Crohn disease | |
Due to inflammation and damage to cartilaginous airways | |
Congenital structural defects | |
Lymphatic | Yellow nail syndrome |
Tracheobronchial | Williams-Campbell syndrome (cartilage deficiency) Tracheobronchomegaly (eg, Mounier-Kuhn syndrome) |
Vascular | Pulmonary sequestration (a congenital malformation in which a nonfunctioning mass of lung tissue lacks normal communication with the tracheobronchial tree and receives its arterial blood supply from the systemic circulation) |
Toxic inhalation | |
Ammonia Chlorine Nitrogen dioxide | Direct airway damage altering structure and function |
Other | |
Transplantation | May be secondary to frequent infection due to immunosuppression |
Diffuse panbronchiolitis | Rare syndrome involving bronchiolitis and chronic sinusitis |
Obstructive lung disease | May occur with advanced chronic obstructive pulmonary disease (COPD) or asthma |
Chronic aspiration | Due to severe gastroesophageal reflux disease or swallowing dysfunction, most commonly in the lower lobes |
Data from Barker, AF: Bronchiectasis. N England J Med 346:1383–1393, 2002 doi: 10.1056/NEJMra012519 and O'Donnell AE: Bronchiectasis - A Clinical Review. N Engl J Med 387(6):533–545, 2022. doi:10.1056/NEJMra2202819. | |
Etiology references
1. Wang L, Wang J, Zhao G, Li J. Prevalence of bronchiectasis in adults: a meta-analysis. BMC Public Health 2024;24(1):2675. doi:10.1186/s12889-024-19956-y
2. Gómez-Olivas JD, Oscullo G, Martínez-García MÁ. Etiology of Bronchiectasis in the World: Data from the Published National and International Registries. J Clin Med 2023;12(18):5782. doi:10.3390/jcm12185782
Pathophysiology of Bronchiectasis
Bronchiectasis represents a common end-point of several disorders predisposing to chronic airway inflammation. The most widely accepted model describes a "vicious cycle" that results in airway damage and dilation (1). An initial insult to the airways (eg, infectious, autoimmune, toxic inhalations, etc) leads to airway inflammation and epithelial damage. This initial damage and the resulting inflammatory response create structural changes in the airways that impair mucociliary clearance and promote bacterial colonization and infection. This process perpetuates, ultimately resulting in irreversible airway damage if the cycle continues untreated (2).
The role of neutrophilic inflammation in the development of bronchiectasis is increasingly recognized. Inflammation of small and medium-sized airways from a causative disorder releases inflammatory mediators such as proteases (specifically, elastase) from intraluminal neutrophils. The inflammatory mediators destroy elastin, cartilage, and muscle in larger airways, resulting in irreversible bronchodilation. Neutrophil extracellular traps (NETs), a web of DNA ejected from neutrophils that are studded with neutrophil granule enzymes and proteins, are a key mechanism underlying chronic and self-perpetuating airway inflammation. Simultaneously, in the inflamed small and medium-sized airways, macrophages and lymphocytes form chronic infiltrates that thicken mucosal layers. This thickening causes the airway obstruction frequently noted during pulmonary function testing.
With disease progression, inflammation spreads beyond the airways, causing fibrosis of the surrounding lung parenchyma. What inflames the small airways depends on the etiology of bronchiectasis. Common contributors include impaired airway clearance (due to the production of thick, viscous mucus in CF, lack of ciliary motility in primary ciliary dyskinesia [PCD], or damage to the cilia and/or airways secondary to infection or injury) and impaired host defenses; these factors predispose patients to chronic infection and inflammation. Atopic inflammation involving IgE can contribute to airway damage in patients with asthma and ABPA. In the case of immune deficiency (particularly CVID) and autoimmune causes, autoimmune inflammation may also contribute.
When the initial insult is localized, for example when a large airway becomes obstructed, the damage may be more focal within the lungs. The resulting inability to clear secretions leads to a similar cycle of infection, inflammation, and airway wall damage as described above. The right middle lobe is involved most often because its bronchus is small, angulated, and has lymph nodes in close proximity. Lymphadenopathy due to mycobacterial infection sometimes causes bronchial obstruction and focal bronchiectasis.
As ongoing inflammation changes airway anatomy, pathogenic bacteria (sometimes including mycobacteria), colonize the airways (3). Common organisms include:
Pseudomonas aeruginosa
Staphylococcus aureus
Haemophilus influenzae
Nontuberculous mycobacteria
Moraxella catarrhalis
Streptococcus pneumoniae
S. aureus colonization is strongly associated with CF; a culture finding of S. aureus should raise concern for undiagnosed CF.
Colonization with P. aeruginosa tends to indicate severe disease and predicts worse outcomes, including increased risk of exacerbations, hospitalization, poor quality of life, rapid decline in lung function, and death.
Colonization by multiple organisms is common, and antibiotic resistance is a concern in patients who require frequent courses of antibiotics for treatment of exacerbations.
Complications
As the disease progresses, chronic inflammation and hypoxemia can cause neovascularization of the bronchial (not the pulmonary) arteries. Bronchial artery walls rupture easily, leading to hemoptysis, which can be massive and life threatening. Other vascular complications include pulmonary hypertension due to vasoconstriction, arteritis, and sometimes shunting from bronchial to pulmonary vessels. Right heart failure and respiratory failure may occur in severe cases.
Colonization with multidrug-resistant organisms can lead to chronic, low grade airway inflammation. This inflammation can become irreversible, causing recurrent exacerbations and worsening airflow limitation on pulmonary function tests.
Inflammation and cytokine excess may contribute to the development of lower lean body mass (4).
Pathophysiology references
1. Cole PJ: Inflammation: a two-edged sword—the model of bronchiectasis. Eur J Respir Dis Suppl 147:6–15, 1986.
2. O'Donnell AE: Bronchiectasis - A Clinical Review. N Engl J Med 387(6):533–545, 2022. doi:10.1056/NEJMra2202819
3. Aksamit TR, O'Donnell AE, Barker A, et al: Adult Patients With Bronchiectasis: A First Look at the US Bronchiectasis Research Registry. Chest 151(5):982–992, 2017. doi:10.1016/j.chest.2016.10.055
4. Ionescu AA, Nixon LS, Evans WD, et al: Bone density, body composition, and inflammatory status in cystic fibrosis. Am J Respir Crit Care Med 162(3 Pt 1):789–794, 2000. doi:10.1164/ajrccm.162.3.9910118
Symptoms and Signs of Bronchiectasis
Symptoms characteristically begin insidiously, gradually worsen over years, and may be accompanied by episodes of acute exacerbation. A small proportion of patients may have no symptoms.
The most common presenting symptom is chronic cough that produces thick, tenacious, and often purulent sputum. Dyspnea and wheezing are common, and pleuritic chest pain can develop. In advanced cases, hypoxemia and right-sided heart failure due to pulmonary hypertension may increase dyspnea. Hemoptysis can occur in some patients due to airway neovascularization and can be massive and life-threatening.
Acute exacerbations can be common and frequently result from new or worsened infection. Exacerbations are marked by a worsening cough and increases in dyspnea and the volume and purulence of sputum. Low-grade fever and constitutional symptoms (eg, fatigue, malaise) may also be present.
Halitosis and abnormal breath sounds, including crackles, rhonchi, and wheezing, are typical physical examination findings. Dullness over parenchymal or airway areas obstructed by mucus may be present on percussion. Digital clubbing is uncommon but may be present, particularly in patients with CF. Chronic rhinosinusitis and nasal polyps may be present, also in patients with CF or PCD. Lean body mass commonly decreases in patients with CF or malabsorption.
Diagnosis of Bronchiectasis
Chest -radiograph
High-resolution chest computed tomography (CT)
Pulmonary function tests for baseline evaluation and monitoring disease progression
Sputum culture for bacteria and mycobacteria to determine colonizing organisms
Specific tests for suspected predisposing diseases
Diagnosis is based on history, physical examination, and radiologic testing, beginning with a chest radiograph. Chronic bronchitis may mimic bronchiectasis clinically, but bronchiectasis is distinguished by increased purulence and volume of daily sputum and by dilated airways shown on imaging studies.
Imaging
Chest radiograph alone may sometimes be diagnostic. Radiography findings suggestive of bronchiectasis involve thickening of the airway walls and/or airway dilation; typical findings include ill-defined linear perihilar densities with indistinctness of the central pulmonary arteries, indistinct rings due to thickened airways seen in cross section (parallel to the x-ray beam), and “tram lines” (or tram-track sign) caused by thickened, dilated airways perpendicular to the x-ray beam. Dilated airways filled with mucous plugs can also cause scattered elongated, tubular opacities.
Radiographic patterns may differ depending on the underlying disease; bronchiectasis due to CF develops predominantly in the upper lobes, whereas bronchiectasis due to an endobronchial obstruction causes more focal radiographic abnormalities.
High-resolution CT is the imaging modality of choice to definitively diagnose as well as define the extent of bronchiectasis. Typical CT findings include airway dilation (in which the inner lumen of 2 or more airways exceeds the diameter of the adjacent artery) and the signet ring sign, in which a thickened, dilated airway appears adjacent to a smaller artery in transaxial view. Lack of normal bronchial tapering can result in visible medium-sized bronchi extending almost to the pleura. "Tram lines" may be easily visible on CT and are indicative of dilated, thickened airway walls (parallel lines) filled with mucus.
As airway damage increases over time, bronchiectasis changes progress from cylindrical to varicose and then cystic findings on imaging. Atelectasis, consolidation, mucous plugs, and decreased vascularity are nonspecific findings.
Traction bronchiectasis occurs due to tension and opening of the airways by adjacent fibrotic lung parenchyma, which is a secondary finding and hallmark of various fibrotic lung diseases including idiopathic pulmonary fibrosis (IPF) or other forms of interstitial lung disease. Traction bronchiectasis is not considered a primary bronchiectasis.

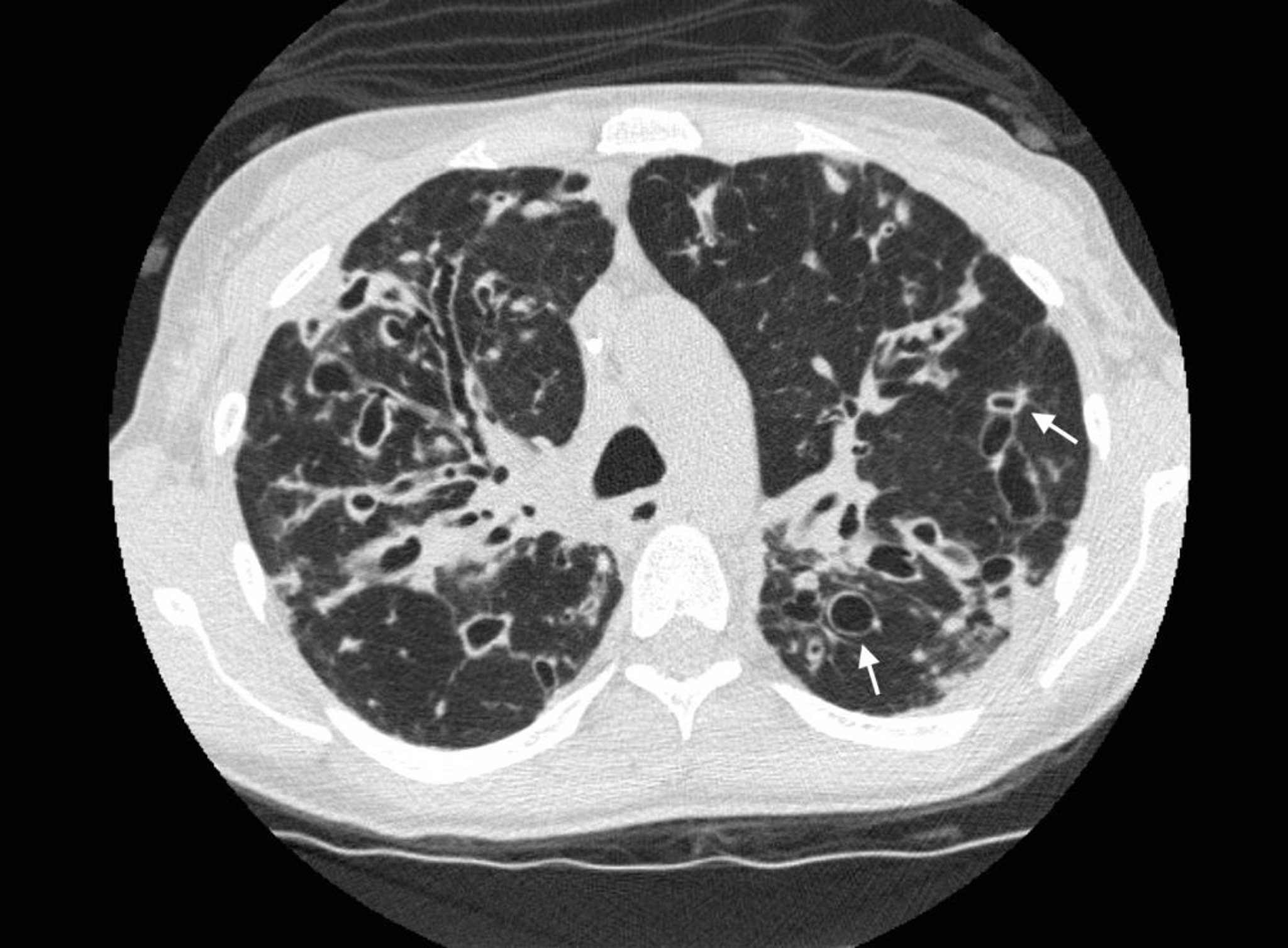
A chest CT in a patient with cystic fibrosis shows severe bronchiectasis in the upper lung zones with dilated airways and cystic changes. Arrows show the signet ring sign, where a dilated airway (the ring) is adjacent to a smaller artery (the top of the ring). Normally, airways are the same size as, or smaller than, the adjacent arteries.
Photo courtesy of Başak Çoruh, MD.

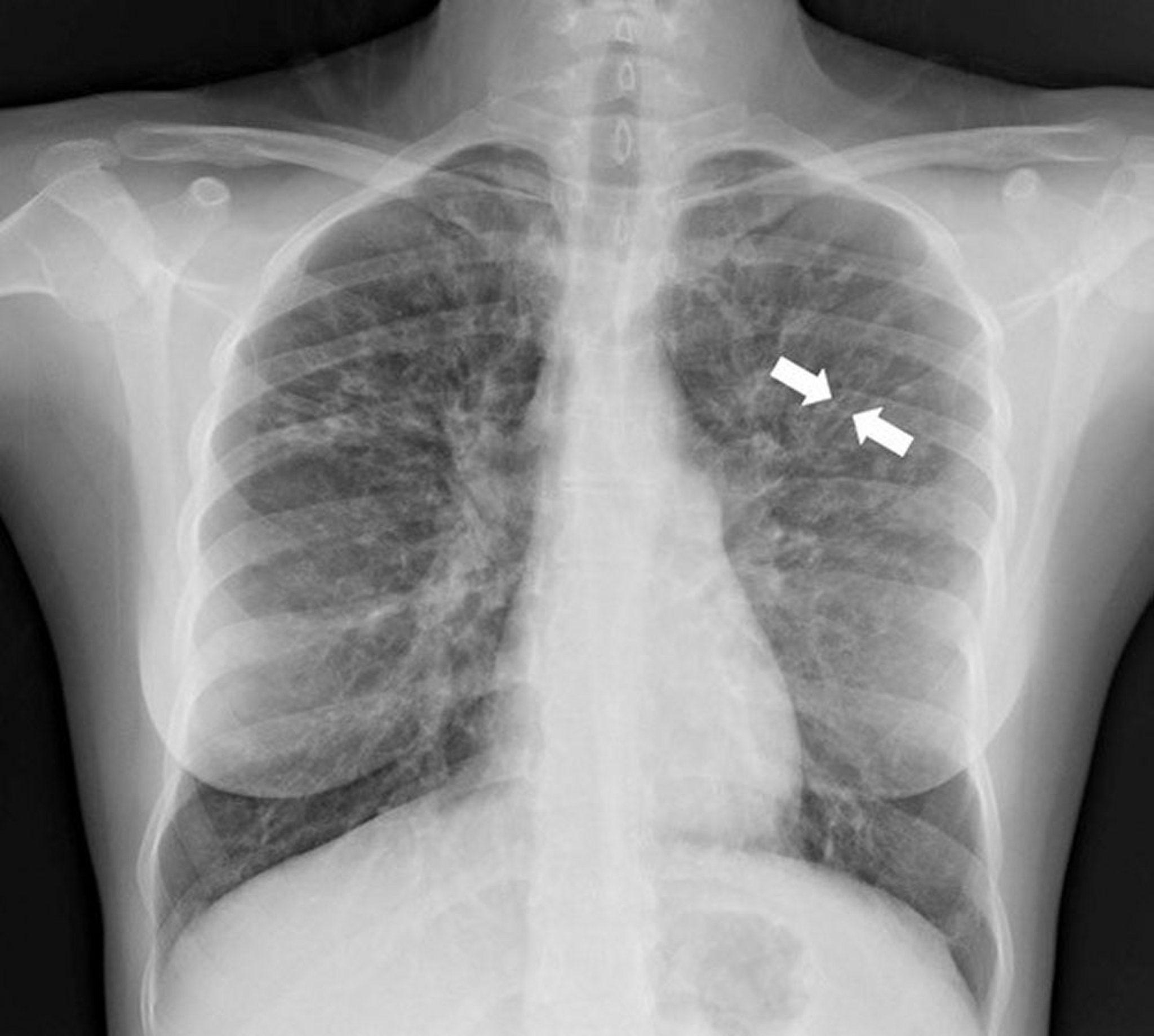
A chest radiograph in a patient with cystic fibrosis demonstrating upper lung zone predominant bronchiectasis. The white arrows are outlining the “tram-track sign,” a dilated airway with thickened walls, sometimes filled with mucus, that are parallel to one another (similar to a tram track).
Photo courtesy of Başak Çoruh, MD.
Pulmonary function tests
Pulmonary function tests (PFTs) can be helpful for documenting baseline function and for monitoring disease progression; however, PFT results must be interpreted in the context of the evolving disease course and overall clinical picture. PFTs may be normal. Airflow obstruction (reduced forced expiratory volume in 1 second [FEV1] with reduction in the FEV1/forced vital capacity [FVC] ratio) is the most common abnormality present in bronchiectasis, with or without a significant response to beta-agonist bronchodilators. FVC may be reduced, either due to air-trapping in the setting of airflow obstruction or the development of parenchymal fibrosis and restriction in more advanced cases. Lung volume measurements can distinguish between these 2 processes. In more advanced disease, decreased diffusing capacity for carbon monoxide (DLCO) may also be observed.
Diagnosis of cause
During an exacerbation-free period, all patients should have expectorated or induced sputum cultured to determine the predominant colonizing bacteria and their sensitivities. This information helps with antibiotic selection during exacerbations.
A complete blood count (CBC) and differential can help determine the severity of disease activity and identify eosinophilia, which may suggest complicating diagnoses such as allergic asthma, or ABPA.
Gram staining and cultures of sputum for bacterial, mycobacterial (Mycobacterium avium complex and M. tuberculosis), and fungal (Aspergillus species) organisms may also help identify infectious causes of chronic airway inflammation.
Clinically significant nontuberculous mycobacterial infection is diagnosed by isolating these organisms in ≥ 2 cultures from serial sputum samples (expectorated or induced) or a single bronchoalveolar lavage sample. Infection can also be diagnosed histopathologically if granulomatous inflammation is present on biopsy in addition to a positive culture.
Additional testing based on the history and imaging findings may be done when the cause of bronchiectasis is unclear. Specific tests to identify the cause of bronchiectasis may include the following:
Alpha-1 antitrypsin level to evaluate for alpha-1 antitrypsin deficiency if high-resolution CT shows lower lobe emphysema
Rheumatoid factor (RF), antinuclear antibody (ANA), and antineutrophil cytoplasmic antibody (ANCA) testing if a systemic rheumatic disease is suspected
Serum immunoglobulins (IgG, IgA, IgM) via serum electrophoresis to diagnose common variable immunodeficiency
Serum IgE levels, allergy skin-testing, and Aspergillus precipitins (IgG) if patients have eosinophilia, to exclude ABPA
Sweat chloride tests (a positive test should be confirmed with a repeat test) and CFTR gene mutation analysis to diagnose CF (including in adults > 40 years without an identifiable cause of bronchiectasis, especially those with upper lobe involvement, malabsorption, or male infertility)
HIV testing to exclude acquired immunodeficiency
Patients who have laboratory evidence of immunodeficiency should be referred to an immunology specialist for evaluation because test results are often challenging to interpret. Additional specialized testing, including quantitative and qualitative analysis of B- and T-cell subsets, IgG subclasses, and serologic responses to vaccines, may also be required to confirm the type of immunodeficiency present and determine treatment options.
Primary ciliary dyskinesia (PCD) should be considered if adults with bronchiectasis also have chronic sinus disease or otitis media, particularly if problems have persisted since childhood. Bronchiectasis in such patients may have right middle lobe and lingular predominance, and infertility in males or dextrocardia on radiography may be present. Nasal or fractional exhaled nitric oxide (FeNO) level is frequently low. Definitive diagnosis requires examination of a nasal or bronchial epithelial sample for abnormal ciliary structure using transmission electron microscopy. The diagnosis of PCD should typically be done in specialized centers because evaluation can be challenging. Nonspecific structural defects can be present in up to 10% of cilia in healthy people and in patients with pulmonary disease, and infection can cause transient dyskinesia. Ciliary ultrastructure may also be normal in some patients with PCD syndromes, requiring further testing to identify abnormal ciliary function.
Bronchoscopy is indicated when an anatomic or obstructive lesion is suspected as the cause of bronchiectasis. It is also used to remove any foreign body that may be present and leading to bronchiectasis. Bronchoscopy is occasionally needed to diagnose infection such as nontuberculous mycobacteria (NTM).
Definition and evaluation of exacerbations
A bronchiectasis exacerbation is defined as a patient with bronchiectasis with deterioration for at least 48 hours and ≥ 3 of the following symptoms (1):
Breathlessness and/or exercise intolerance
Cough
Fatigue and/or malaise
Hemoptysis
Increase in sputum purulence
Increase in sputum volume and/or consistency
In addition, a clinician must determine that a change in treatment is warranted.
Once an exacerbation has been identified, testing should include sputum culture and Gram stain to identify any causative organisms and determine antimicrobial sensitivity patterns. Pulse oximetry should be measured if there is concern for hypoxemia. Chest radiography should be obtained in patients with fever, chills, hypoxemia, or more severe dyspnea to evaluate superimposed pneumonia or exclude complications such as a lung abscess and empyema. More severe systemic symptoms or signs of sepsis should prompt laboratory testing such as a CBC, renal function and electrolytes, arterial blood gas, and serum lactate.
Diagnosis reference
1. Hill AT, Haworth CS, Aliberti S, et al: Pulmonary exacerbation in adults with bronchiectasis: A consensus definition for clinical research. Eur Respir J 49:1700051, 2017.
Treatment of Bronchiectasis
Prevention of exacerbations with regular vaccinations and sometimes suppressive antibiotics
Measures to help clear airway secretions
Bronchodilators in patients with airflow obstruction and/or significant dyspnea
Inhaled corticosteroids if complicating asthma or ABPA
Antibiotics and bronchodilators for acute exacerbations
Early treatment with antivirals of any viral infections, particularly influenza and COVID-19
Sometimes surgical resection for localized disease with intractable symptoms or bleeding
Lung transplantation in carefully selected patients who have advanced disease despite maximal therapy
The key treatment goals are to control symptoms and improve quality of life, reduce the frequency of exacerbations, and preserve lung function (1).
As for all patients with chronic pulmonary disease, recommendations include the following:
Referral to pulmonary rehabilitation
Annual influenza vaccination
Respiratory syncytial virus vaccination as clinically indicated
Pulmonary rehabilitation is recommended for patients with bronchiectasis to improve exercise capacity and health-related quality of life, and to reduce symptoms such as dyspnea and fatigue (2). Patients can also be instructed and coached on performing airway clearance during rehabilitation sessions.
Airway clearance techniques are key components of treatment that aim to reduce chronic cough in patients with significant sputum production and mucous plugging and to reduce symptoms during exacerbations. Such techniques include regular exercise, chest physiotherapy with postural drainage and chest percussion, positive expiratory pressure devices, intrapulmonary percussive ventilators, pneumatic vests, and autogenic drainage or active cycle of breathing (breathing techniques thought to help move secretions from peripheral to central airways, thus facilitating expectoration). Patients should be taught these techniques by a respiratory therapist and should use whichever one is most effective and sustainable for them; no evidence favors one particular technique. Patients should be advised to continue their airway clearance techniques for at least 10 minutes and may stop when they produce 2 clear coughs or huffs (3) or 30 minutes has passed.
Airway clearance sessions for bronchiectasis should be done in the following order, depending on which agents are prescribed:
Inhaled short-acting bronchodilators
Mucolytic therapy (if prescribed)
Airway clearance technique
Any prescribed inhaled or nebulized antibiotics, long-acting bronchodilators, or corticosteroids
For patients with airway obstruction, bronchodilator therapy (eg, with some combination of a long-acting beta-adrenergic agonist [LABA] and/or a long-acting muscarinic antagonist [LAMA]) may be used as indicated by symptoms and severity of lung obstruction, similar to the use in patients with COPD. Limited evidence suggests bronchodilator therapy can help improve lung function (4). Inhaled corticosteroids (ICS) may be used in some patients, specifically in those with coexisting asthma, but may increase risk of infection in other patients without clear indications (1). Short-acting beta-adrenergic agonists (SABA) such as albuterol are used to relieve acute airway obstruction causing wheezing or dyspnea. In some patients with eosinophilic airway inflammation, such as those with asthma, biologic therapies targeting Th2 T-helper cell induced IgE-mediated responses and eosinophil activity (eg, omalizumab, mepolizumab, dupilumab) may be required for long-term maintenance therapy. Maintaining adequate hydration is also important.
Patients with cystic fibrosis may receive nebulized treatments, including a mucolytic (rhDNase, also called dornase alfa) and hypertonic (7%) saline, to help reduce sputum viscosity and enhance airway clearance. In patients without CF, evidence of benefit with these measures is inconclusive, so only humidification and saline are recommended as inhaled treatments. Inhaled rhDNase may be harmful in patients with bronchiectasis not caused by CF.
Use of suppressive antibiotics regularly or on a rotating schedule reduces symptoms and exacerbations but may increase the risk that future infections will involve resistant organisms. Guidelines suggest using antibiotics in patients with ≥ 3 exacerbations per year and possibly also in those with fewer exacerbations who have culture-proven P. aeruginosa colonization. Some guidelines suggest attempting eradication of organisms such as P. aeruginosa or S. aureus when they are first detected in sputum cultures (3).
Chronic macrolide therapy (eg, oral azithromycin given once daily or 3 times a week) reduces acute exacerbations in patients with bronchiectasis, and can slow the decline in lung function in patients with CF (5). Macrolides are thought to be beneficial mainly due to their anti-inflammatory or immunomodulatory effects. Nebulized antibiotics (eg, amikacin, aztreonam, ciprofloxacin, gentamicin, tobramycin) can reduce sputum bacterial load, and may also reduce the frequency of exacerbations. The evidence supporting their use and benefit is strongest in patients with CF.
Most patients with bronchiectasis who develop influenza or SARS-CoV-2 infection should be treated with antiviral medications to prevent complications and development of severe disease. For influenza, treatment is typically with oseltamivir regardless of the duration of viral symptoms. SARS-CoV-2 infections should be treated as soon as possible in patients with bronchiectasis, especially in those with underlying immune suppression. The treatment approach is individualized, depending on the severity of infection and other patient risk factors. Oral corticosteroids are not recommended for outpatients with COVID-19 in the absence of a co-existing condition, such as asthma or COPD, that is known to benefit from corticosteroids. Patients with bronchiectasis should be monitored closely for bacterial co-infection, and any exacerbation triggered by viral infection should be treated.
Underlying conditions should be treated to slow the progression of lung disease.
For patients with underlying immunodeficiency states: Scheduled intravenous or subcutaneous immunoglobulin replacement therapies (which reduce the frequency of lower respiratory infections [6])
For patients with cystic fibrosis: Antibiotics and inhaled bronchodilators as well as comprehensive support, and dietary supplementation. Most patients with cystic fibrosis benefit from CFTR modulator therapy, which can decrease exacerbations. Patients with CF should receive all or part of their care by teams with expertise in CF, generally at a designated CF care center.
For patients with allergic bronchopulmonary aspergillosis: Corticosteroids and sometimes azole antifungals.
Biologic agents may be used to target type 2 (Th2-high) asthma with eosinophilic airway inflammation.
For patients with alpha-1 antitrypsin deficiency: Replacement therapy for those who qualify based on serum levels and FEV1.
Approaches that have shown promise are under investigation for treatment of bronchiectasis.
Acute exacerbations
Acute exacerbations are treated with antibiotics, inhaled bronchodilators (particularly if patients are dyspneic or wheezing), increased attempts at mucus clearance using mechanical techniques, treatment of dehydration (if present), humidification, and nebulized saline (and mucolytics for patients with CF). Systemic corticosteroids should not routinely be used to treat exacerbations except in patients with COPD or asthma. Antibiotic choice depends on previous culture results and whether or not patients have CF (7).
Initial antibiotics for patients without CF and with no prior culture results should be effective against H. influenzae, M. catarrhalis, S. aureus, and S. pneumoniae. Examples include amoxicillin/clavulanic acid, azithromycin, clarithromycin, and doxycycline. Patients with known P. aeruginosa colonization or more severe exacerbations should receive antibiotics effective against this organism (eg, ciprofloxacin, levofloxacin) until repeat culture results are available. Antibiotics should be adjusted based on culture results and are given for a typical duration of 14 days, especially if P. aeruginosa is detected. Shorter courses are reserved for patients with mild disease.
Initial antibiotic selection for patients with CF is guided by previous sputum culture results (done routinely in all patients with CF). During childhood, common infecting organisms are S. aureus and H. influenzae, and quinolone antibiotics such as ciprofloxacin and levofloxacin may be used. In the later stages of CF, infections involve highly resistant strains of certain gram-negative organisms including P. aeruginosa, Burkholderia cepacia, and Stenotrophomonas maltophilia. In patients with infections caused by these organisms, treatment is with multiple antibiotics (eg, tobramycin, ceftazidime, cefepime, piperacillin/tazobactam, meropenem). Intravenous administration is frequently required.
Complications
Significant hemoptysis is usually treated with bronchial artery embolization, but surgical resection may be considered if embolization is ineffective and pulmonary function is adequate.
Colonization with mycobacterial organisms such as M. avium complex almost always requires multiple antibiotic regimens that include clarithromycin or azithromycin; rifampin or rifabutin; and ethambutol. Antibiotic therapy is typically continued until sputum cultures, repeated every 1 to 2 months, have been negative for 12 months. More complex regimens with multiple medications may be required for patients with drug-resistant mycobacteria or difficult-to-clear infections.
Surgical resection is rarely indicated but may be considered when bronchiectasis is focal or localized, medical therapy has been optimized, and the symptoms are intolerable. In certain patients with diffuse bronchiectasis, especially cystic fibrosis, lung transplantation is also an option.
Treatment references
1. O'Donnell AE: Bronchiectasis - A Clinical Review. N Engl J Med 387(6):533–545, 2022. doi:10.1056/NEJMra2202819
2. Rochester CL, Alison JA, Carlin B, et al: Pulmonary Rehabilitation for Adults with Chronic Respiratory Disease: An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med 208(4):e7–e26, 2023. doi:10.1164/rccm.202306-1066ST
3. Hill AT, Sullivan AL, Chalmers JD, et al: British Thoracic Society Guideline for bronchiectasis in adults. Thorax 74(Suppl 1):1–69, 2019. doi: 10.1136/thoraxjnl-2018-212463
4. Jeong HJ, Lee H, Carriere KC, et al: Effects of long-term bronchodilators in bronchiectasis patients with airflow limitation based on bronchodilator response at baseline. Int J Chron Obstruct Pulmon Dis 11:2757–2764, 2016. doi:10.2147/COPD.S115581
5. Chalmers JD, Boersma W, Lonergan M, et al. Long-term macrolide antibiotics for the treatment of bronchiectasis in adults: an individual participant data meta-analysis. Lancet Respir Med 7(10):845–854, 2019. doi:10.1016/S2213-2600(19)30191-2
6. Quinti I, Sorellina A, Guerra A, et al: Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: Results from a multicenter prospective cohort trial. J Clin Immunol 31: 315–322, 2011.
7. Flume PA, Mogayzel PJ Jr, Robinson KA, et al: Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am J Respir Crit Care Med 80:802–808, 2009. doi: 10.1164/rccm.200812-1845PP
Prognosis for Bronchiectasis
Prognosis varies widely. In the United States, the overall all-cause mortality rate at 5 years for patients with any kind of bronchiectasis was 12.1%, indicating a generally favorable prognosis (1). Mean yearly decrease in FEV1 is about 50 to 55 mL (normal decrease in healthy people is about 20 to 30 mL).
Patients with CF historically have had the poorest prognosis, with a median survival of 36 years. However, the advent of CFTR (cystic fibrosis transmembrane regulator) modulator therapy has resulted in meaningful improvements in outcomes, even in patients with advanced lung disease (2).
Prognosis references
1. Aksamit TR, Locantore N, Addrizzo-Harris D, et al: Five-Year Outcomes among U.S. Bronchiectasis and NTM Research Registry Patients. Am J Respir Crit Care Med 210(1):108–118, 2024. doi:10.1164/rccm.202307-1165OC
2. Shteinberg M, Taylor-Cousar JL: Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur Respir Rev 29(155):190112, 2020. doi: 10.1183/16000617.0112-2019
Key Points
In bronchiectasis, chronic inflammation from various causes destroys elastin, cartilage, and muscle in larger airways, resulting in irreversible damage and dilated airways that become chronically colonized by infectious organisms.
Patients have chronic productive cough with intermittent acute exacerbations.
Diagnosis is with imaging, usually CT; cultures should be done to identify colonizing organism(s).
Prevent exacerbations using appropriate immunizations, airway clearance measures, and sometimes macrolide antibiotics.
Treat exacerbations with antibiotics, bronchodilators, more frequent airway clearance measures, and sometimes corticosteroids.
Drug Information for the Topic





