

Fabry disease is a type of lysosomal storage disorder called a sphingolipidosis. It is caused by a buildup of glycolipid in tissues. This disease causes skin growths, pain in the extremities, poor vision, recurring episodes of fever, kidney failure, and heart disease. Fabry disease occurs when parents pass on to their children a defective gene that causes this disease.
Fabry disease occurs when the body lacks enzymes needed to break down a glycolipid.
Symptoms include skin growths, eye problems, kidney failure, and heart disease.
The diagnosis is based on the results of prenatal screening tests, newborn screening tests, and other blood tests.
Treatment includes enzyme replacement therapy.
Most children who have Fabry disease live to adulthood.
There are different types of inherited disorders. In many hereditary metabolic disorders, usually 2 copies of the abnormal gene are necessary for the disorder to occur, so an affected child inherited the abnormal gene from both parents. (Often neither parent has the disorder.) Some hereditary metabolic disorders are X-linked, which means, for boys, inheriting only 1 copy of the abnormal gene can cause the disorder. (See also Overview of Hereditary Metabolic Disorders.)
Sphingolipidoses occur when people do not have the enzymes needed to break down (metabolize) sphingolipids, which are compounds that protect the cell surface and serve certain functions in the cells. There are many types of sphingolipidoses besides Fabry disease.
In Fabry disease, a glycolipid, which is a product of fat metabolism, accumulates in tissues. The enzyme needed to breakdown the glycolipid, called alpha-galactosidase A, does not work correctly. Because the defective gene for this rare inherited disorder is carried on the X chromosome, the full-blown disease occurs only in boys (see X-Linked Inheritance). Because girls have 2 X chromosomes, affected girls may have symptoms but do not develop full-blown Fabry disease.

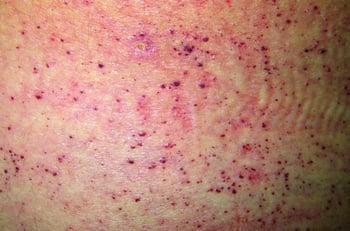
This photo shows noncancerous (benign) skin growths (angiokeratomas) on the abdomen of a person who has Fabry disease.
The accumulation of glycolipid causes noncancerous (benign) skin growths (angiokeratomas) to form on the lower part of the trunk. The corneas become cloudy, resulting in poor vision. A burning pain may develop in the arms and legs, and children may have episodes of fever.
Diagnosis of Fabry Disease
Prenatal screening tests
Newborn screening tests
Other blood tests
Before birth, Fabry disease can be diagnosed in the fetus by using the prenatal screening tests chorionic villus sampling or amniocentesis.
After birth, Fabry disease may be diagnosed in some states in the United States by routine newborn screening tests.
Doctors measure levels of alpha-galactosidase A in blood or in white blood cells.
Treatment of Fabry Disease
Enzyme replacement therapy
Sometimes kidney transplant
Doctors treat Fabry disease with enzyme replacement therapy (agalsidase beta). Treatment also consists of taking medications to help relieve pain and fever.Doctors treat Fabry disease with enzyme replacement therapy (agalsidase beta). Treatment also consists of taking medications to help relieve pain and fever.
People with kidney failure may need a kidney transplant.
Prognosis for Fabry Disease
Children with Fabry disease eventually develop kidney failure and heart disease, although, most often, they live to adulthood. Kidney failure may lead to high blood pressure, which may result in stroke.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
National Organization for Rare Disorders (NORD): This resource provides information to parents and families about rare diseases, including a list of rare diseases, support groups, and clinical trial resources.
Genetic and Rare Diseases Information Center (GARD): This resource provides and easy to understand information about rare or genetic diseases.
Drug Information for the Topic





