

A number of systemic rheumatic diseases cause inflammation of the uveal tract. (See also Overview of Uveitis.)
Spondyloarthropathies
The seronegative spondyloarthropathies are a common cause of anterior uveitis.
Among the seronegative spondyloarthropathies, ocular inflammation is most common with ankylosing spondylitis (1) but also occurs with reactive arthritis, inflammatory bowel disease (ulcerative colitis and Crohn disease), and psoriatic arthritis. Uveitis is classically unilateral, but recurrences are common and active inflammation may alternate between eyes. Men are affected more commonly than women. Most patients, regardless of sex, are HLA-B27 positive.
Treatment requires a topical corticosteroid and a cycloplegic-mydriatic medication (2). Occasionally, periocular corticosteroid injections are required. Severe chronic cases may require noncorticosteroid immunosuppressive medications (eg, methotrexate, mycophenolate mofetil, tumor necrosis factor inhibitors).). Occasionally, periocular corticosteroid injections are required. Severe chronic cases may require noncorticosteroid immunosuppressive medications (eg, methotrexate, mycophenolate mofetil, tumor necrosis factor inhibitors).
Juvenile idiopathic arthritis (JIA)
JIA, previously known as juvenile RA, characteristically causes chronic bilateral iridocyclitis in children, particularly those with the oligoarticular variety. Unlike most forms of anterior uveitis, however, JIA-associated uveitis tends not to cause pain, photophobia, and conjunctival injection but only blurring and miosis and is, therefore, often referred to as white iritis. It can be asymptomatic. Because symptoms can be overlooked or absent, patients with JIA should be regularly screened.
Rheumatoid arthritis, in contrast, is not associated with isolated uveitis but can cause scleritis, which may cause secondary uveal tract inflammation.
Recurrent bouts of inflammation are best treated with a topical corticosteroid and a cycloplegic-mydriatic medication (3). Given the chronic nature of disease and risk of treatment-related cataract and glaucoma development, long-term control often requires use of a noncorticosteroid immunosuppressive medication (eg, methotrexate, mycophenolate mofetil, tumor necrosis factor inhibitors).development, long-term control often requires use of a noncorticosteroid immunosuppressive medication (eg, methotrexate, mycophenolate mofetil, tumor necrosis factor inhibitors).
Sarcoidosis
Sarcoidosis accounts for up to 25% of cases of uveitis (4), and various studies found that between 10 and 50% of Caucasian patients with sarcoidosis develop uveitis (5) . Sarcoid uveitis is more common among patients of African descent (5) and older patients.
Virtually any symptoms and signs of anterior, intermediate, posterior, or panuveitis can occur. Suggestive findings include conjunctival granulomas; large keratic precipitates on the corneal endothelium (so-called granulomatous or mutton fat precipitates); iris, retinal, or choroidal granulomas; and retinal vasculitis. Biopsy of suggestive lesions, which provides the most secure diagnosis, is usually done on the conjunctiva; it is rarely done on intraocular tissues because of the risk associated with the procedure.
Treatment usually involves topical, periocular (injected), intraocular, or systemic corticosteroids, or a combination, along with a topical cycloplegic-mydriatic medication (5). Patients with moderate to severe inflammation or chronic disease, or those with systemic involvement, may require a noncorticosteroid immunosuppressive medication (eg, methotrexate, mycophenolate mofetil, tumor necrosis factor inhibitors).). Patients with moderate to severe inflammation or chronic disease, or those with systemic involvement, may require a noncorticosteroid immunosuppressive medication (eg, methotrexate, mycophenolate mofetil, tumor necrosis factor inhibitors).

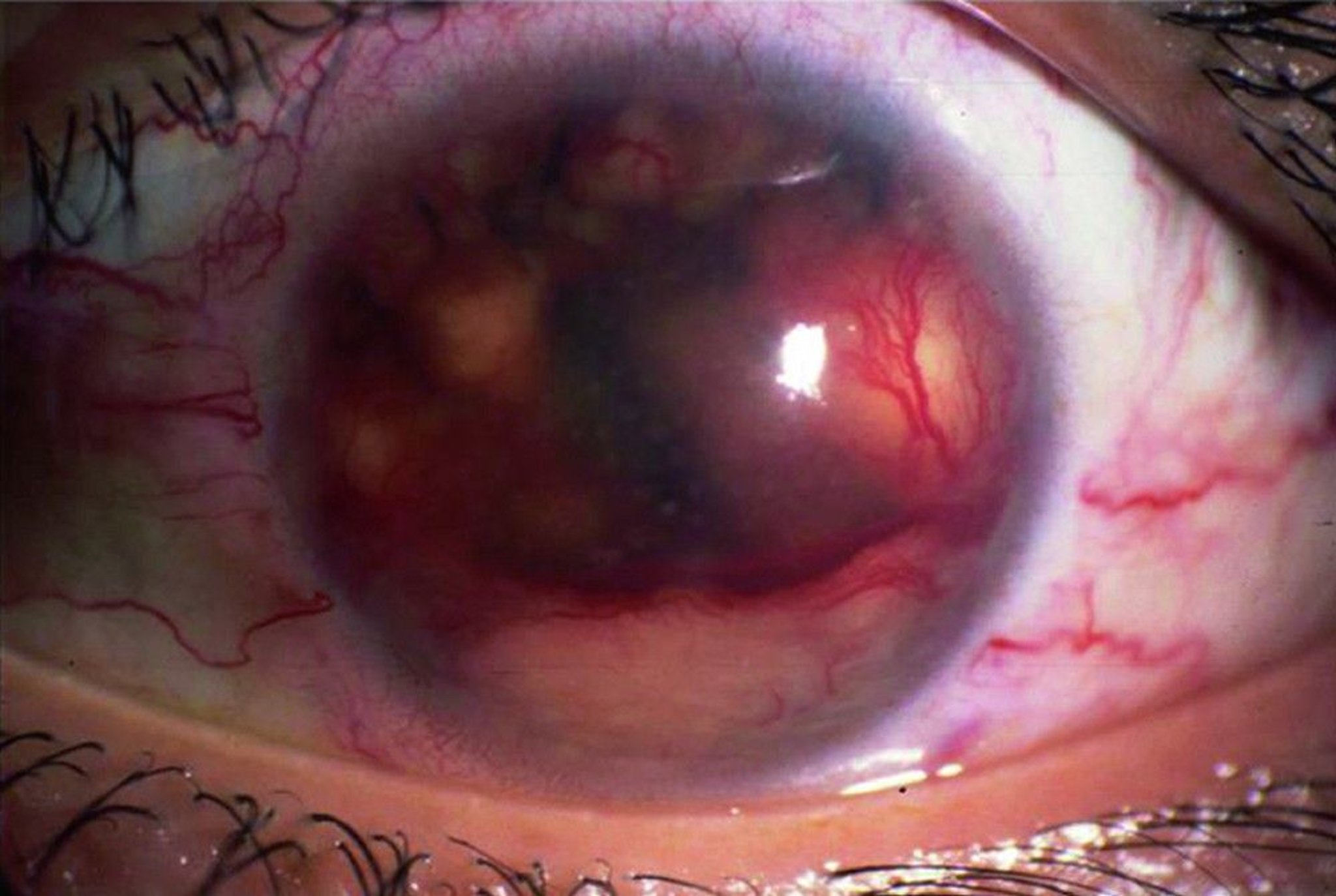
This photo shows iris granulomas (visible as white spots), which are also called papillary margin nodules or Koeppe nodules.
© Springer Science+Business Media
Behçet disease
Behçet disease is rare in North America but is a more common cause of uveitis in countries that extend from the Mediterranean basin to eastern Asia, known as the ancient Silk Road.
Typical findings include severe anterior uveitis with hypopyon, retinitis, retinal vasculitis, and optic disk inflammation. The clinical course is usually severe with multiple recurrences.
Diagnosis requires the presence of associated systemic manifestations, such as oral aphthous or genital ulcers; dermatitis, including erythema nodosum; thrombophlebitis; or epididymitis. Oral aphthae may be biopsied to show an occlusive vasculitis. There are no laboratory tests for Behçet disease, but it is associated with HLA-B51.
Treatment with local and systemic corticosteroids and a cycloplegic-mydriatic medication may alleviate acute exacerbations, but most patients eventually require systemic corticosteroids and a noncorticosteroid immunosuppressive medication to control the inflammation and avoid the serious complications of long-term corticosteroid treatment (6). Biologic agents such as interferons and tumor necrosis factor inhibitors have been effective in selected patients unresponsive to other therapies. Alkylating agents (eg, cyclosporine and chlorambucil) have induced remission.). Biologic agents such as interferons and tumor necrosis factor inhibitors have been effective in selected patients unresponsive to other therapies. Alkylating agents (eg, cyclosporine and chlorambucil) have induced remission.

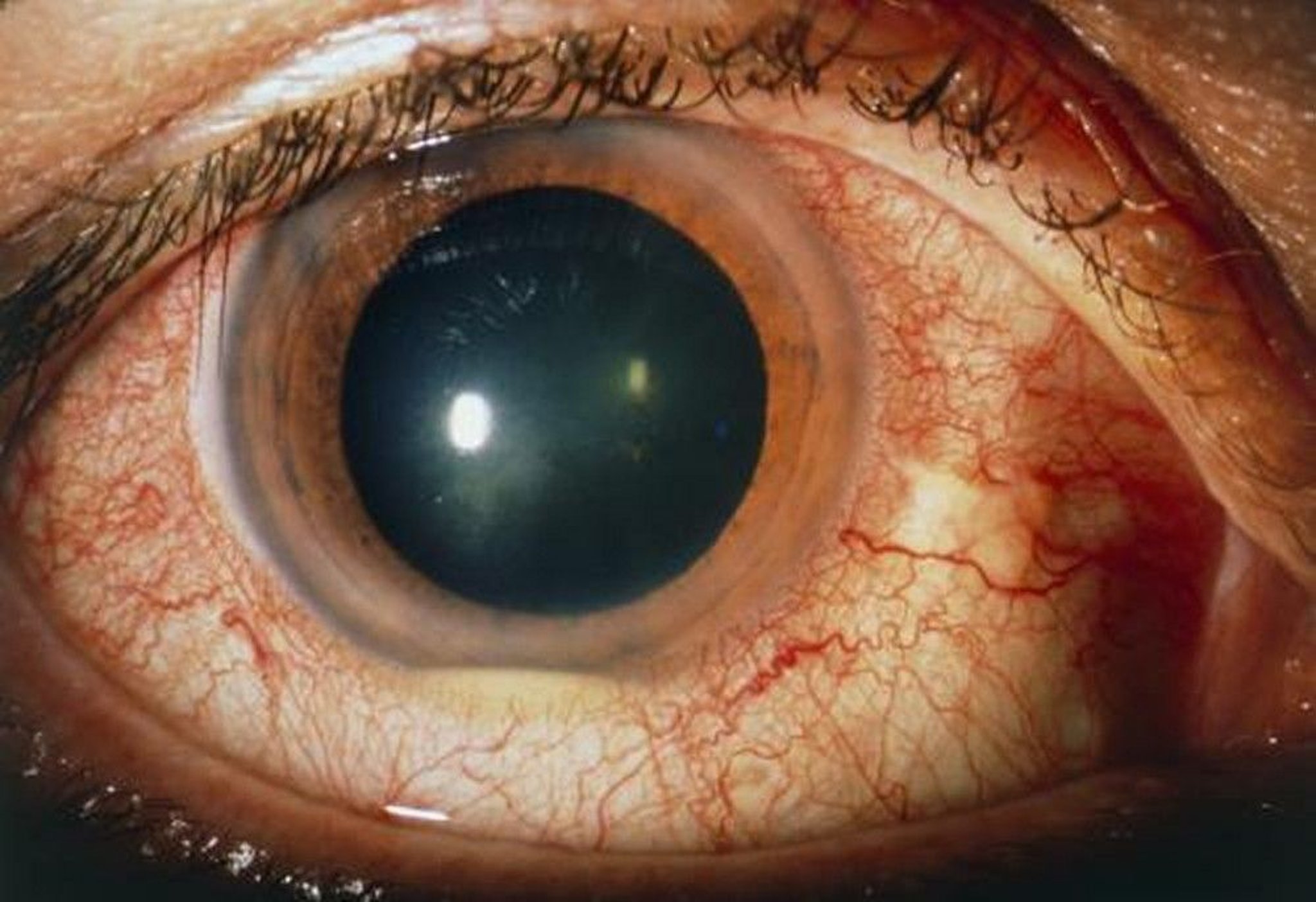
This photo shows pus (or hypopyon) in the anterior chamber of the eye in a patient with Behçet disease. A fine sediment of white blood cells is seen at the bottom of the iris (at lower center) between the iris and cornea. The pupil is enlarged, and the sclera is red with blood vessels.
SUE FORD/SCIENCE PHOTO LIBRARY
Vogt-Koyanagi-Harada (VKH) disease
Vogt-Koyanagi-Harada disease is an uncommon systemic disorder characterized by uveitis accompanied by cutaneous and neurologic abnormalities. VKH disease is particularly common among people of Asian, Asian Indian, American Indian, and Hispanic descent. The etiology is unknown, although an autoimmune reaction directed against melanin-containing cells in the uveal tract, skin, inner ear, and meninges is strongly suspected.
Neurologic symptoms tend to occur early and include tinnitus, dysacusis (auditory agnosia), vertigo, headache, and meningismus. Cutaneous findings frequently occur later and include patchy vitiligo (especially common on the eyelids, low back, and buttocks), poliosis (a localized patch of white hair, which may involve the eyelashes), and alopecia, often involving the head and neck. Common findings include serous retinal detachment, optic disk edema, and choroiditis. Long-term complications include cataracts, glaucoma, subretinal fibrosis, and choroidal neovascularization.
Early treatment includes local and systemic corticosteroids and a cycloplegic-mydriatic medication. Many patients also require noncorticosteroid immunosuppressive medications (eg, methotrexate, mycophenolate mofetil) (Early treatment includes local and systemic corticosteroids and a cycloplegic-mydriatic medication. Many patients also require noncorticosteroid immunosuppressive medications (eg, methotrexate, mycophenolate mofetil) (7).
Tubulointerstitial nephritis and uveitis syndrome (TINU)
TINU typically presents with nongranulomatous acute bilateral anterior uveitis, though it is frequently accompanied by posterior complications, including edema of the optic nerve and macula. It most commonly affects adolescent females but can present at any age. Symptoms include eye pain and redness, decreased visual acuity, and photophobia. Patients may have a history of viral prodrome symptoms and also flank pain, polyuria, and nocturia. Evaluation for nephritis may include urine beta-2 microglobulin levels and sometimes renal biopsy. While the acute kidney injury is frequently self-limited in TINU, the ocular disease is often chronic (8).
Treatment with oral steroids in the acute phase and use of noncorticosteroid immunosuppressive medications (eg, mycophenolate mofetil) for chronic disease is often necessary.Treatment with oral steroids in the acute phase and use of noncorticosteroid immunosuppressive medications (eg, mycophenolate mofetil) for chronic disease is often necessary.
References
1. Rosenbaum JT, Dick AD: The eyes have it: A rheumatologist's view of uveitis. Arthritis Rheumatol 70(10):1533-1543, 2018. doi: 10.1002/art.40568
2. Rademacher J, Poddubnyy D, Pleyer U: Uveitis in spondyloarthritis. Ther Adv Musculoskelet Dis 12:1759720X20951733, 2020. doi: 10.1177/1759720X20951733
3. Oray M, Tugal-Tutkun I: Treatment of juvenile idiopathy arthritis-associated uveitis. Turkish J Ophthalmol 46;2:77-82, 2016. doi: 10.4274/tjo.09581
4. Merrill PT, Kim J, Cox TA, et al: Uveitis in the southeastern United States. Curr Eye Res 16(9):865-874, 1997. doi: 10.1076/ceyr.16.9.865.5048
5. Giorgiutti S, Jacquot R, El Jammal T, et al: Sarcoidosis-related uveitis: A review. J Clin Med 12(9):3194, 2023. doi: 10.3390/jcm12093194
6. Joubert M, Desbois AC, Domont F, et al: Behçet's disease uveitis. J Clin Med 12(11):3648, 2023. doi: 10.3390/jcm12113648.
7. Joye A, Suhler E: Vogt-Koyanagi-Harada disease. Curr Opin Ophthalmol 32(6):574-582, 2021. doi: 10.1097/ICU.0000000000000809.
8. Koreishi AF, Zhou M, Goldstein DA: Tubulointerstitial nephritis and uveitis syndrome: Characterization of clinical features. Ocul Immunol Inflamm 17;29(7-8):1312-1317, 2021. doi: 10.1080/09273948.2020.1736311
Drug Information for the Topic





