

Burkitt lymphoma is an aggressive B-cell non-Hodgkin lymphoma occurring in children and adults. Endemic, sporadic, and immunodeficiency-related forms exist.
(See also Overview of Lymphoma and Non-Hodgkin Lymphomas.)
Classic Burkitt lymphoma is endemic in central Africa and constitutes 30% of childhood lymphomas in the United States. The form endemic to Africa often manifests as enlargement of the jaw or facial bones.


This photo shows a large tumor of the jaw in a patient with Burkitt lymphoma.
M.A. ANSARY/SCIENCE PHOTO LIBRARY
In sporadic Burkitt lymphoma, abdominal disease predominates, often arising in the region of the ileocecal valve or the mesentery. Tumor may cause bowel obstruction. Extranodal sites such as the brain or other solid organs may also be involved. In adults, disease may be bulky and generalized, often with massive involvement of liver, spleen, and bone marrow. Central nervous system (CNS) involvement is often present at diagnosis or with relapsing lymphoma.
Immunodeficiency-related Burkitt lymphoma mostly occurs in patients with human immunodeficiency virus (HIV) infection and less commonly in patients who are bone marrow or solid organ transplant recipients or have other causes of immunodeficiency. In patients with HIV infection, Burkitt lymphoma is considered an AIDS-defining cancer.

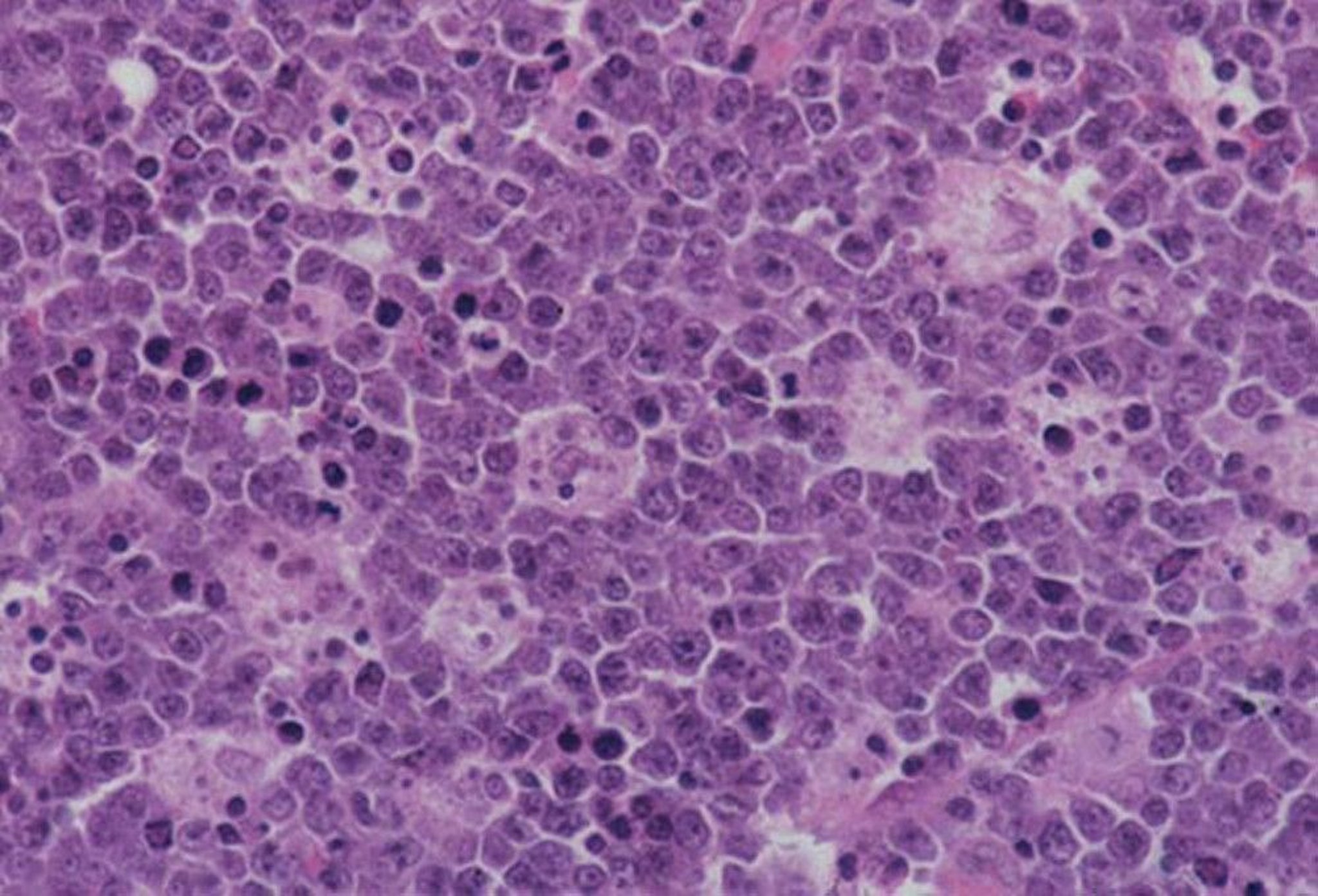
As in patients who do not have an immunodeficiency, Burkitt lymphoma has a "starry-sky" appearance due to the numerous debris-containing macrophages (denoting rapid cell turnover). The tumor also has a high mitotic rate.
By permission of the publisher. From Banks P, et al. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004. Available at www.images.md.
Burkitt lymphoma is the most rapidly growing human tumor, and pathology reveals a high mitotic rate, a monoclonal proliferation of B cells, and a “starry-sky” pattern of benign macrophages that have engulfed apoptotic malignant lymphocytes. On FDG-PET (fluorodeoxyglucose-positron emission tomography) scans, the tumors are highly metabolic. There is a distinctive genetic translocation involving the c-MYC gene on chromosome 8 and the immunoglobulin heavy chain of chromosome 14.
The disease is closely associated with Epstein-Barr virus infection in endemic lymphoma; however, it is uncertain whether Epstein-Barr virus plays an etiologic role.
Diagnosis of Burkitt Lymphoma
Lymph node or bone marrow biopsy
Rarely, laparoscopy
Histopathologic diagnosis is based on biopsy of lymph node or tissue from another suspected disease site, such as the bone marrow. Rarely, laparoscopy may be used for both diagnosis and treatment.
Staging studies must be expedited because the tumor grows rapidly. Staging includes fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT tumor imaging; if unavailable, CT of the chest, abdomen, and pelvis may be done instead.
Patients should also have bone marrow biopsy, cerebrospinal fluid cytology, and laboratory studies, including lactate dehydrogenase (LDH).
Treatment of Burkitt Lymphoma
Intensive chemotherapy
Treatment must be initiated rapidly because these tumors grow rapidly. An intensive alternating regimen of cyclophosphamide, vincristine, doxorubicin, methotrexate, ifosfamide, etoposide, and cytarabine (CODOX-M/IVAC) plus rituximab results is used with success for children and adults Treatment must be initiated rapidly because these tumors grow rapidly. An intensive alternating regimen of cyclophosphamide, vincristine, doxorubicin, methotrexate, ifosfamide, etoposide, and cytarabine (CODOX-M/IVAC) plus rituximab results is used with success for children and adults< 60 years (1). For selected patients < 60 years of age and for many patients > 60 years, regimens such as rituximab plus etoposide, prednisone, vincristine (Oncovin), and doxorubicin (dose-adjusted R-EPOCH) are also commonly used with success. For patients without CNS involvement, CNS prophylaxis (eg, with systemic and/or intrathecal methotrexate and/or cytarabine) is essential. 60 years of age and for many patients > 60 years, regimens such as rituximab plus etoposide, prednisone, vincristine (Oncovin), and doxorubicin (dose-adjusted R-EPOCH) are also commonly used with success. For patients without CNS involvement, CNS prophylaxis (eg, with systemic and/or intrathecal methotrexate and/or cytarabine) is essential.
With treatment, tumor lysis syndrome is common, and patients must receive IV hydration, allopurinol often with rasburicase, alkalinization of the urine (in the absence of hyperphosphatemia), and close attention to electrolytes (particularly potassium, phosphorus, and calcium). is common, and patients must receive IV hydration, allopurinol often with rasburicase, alkalinization of the urine (in the absence of hyperphosphatemia), and close attention to electrolytes (particularly potassium, phosphorus, and calcium).Rasburicase is contraindicated in patients with G6PD deficiency because it may cause hemolytic anemia in these patients. Some patients may require dialysis for hyperkalemia.
If the patient presents with bowel obstruction secondary to tumor but the tumor is completely resected at initial diagnostic-therapeutic laparotomy, then aggressive therapy is still indicated, but fewer cycles may be needed. Patients at the end of treatment should have a complete metabolic response documented by PET or a complete response documented by CT scan and bone marrow biopsy.
Outcome is poor in patients in whom induction fails or relapse (typically within the first 12 months) occurs. Salvage therapy or clinical trials should be considered.
Treatment reference
1. Chamuleau MED, Stenner F, Chitu DA, et al. R-CODOX-M/R-IVAC versus DA-EPOCH-R in patients with newly diagnosed Burkitt lymphoma (HOVON/SAKK): final results of a multicentre, phase 3, open-label, randomised trial. Lancet Haematol 2023;10(12):e966-e975. doi:10.1016/S2352-3026(23)00279-X
More Information
The following English language resource provides information for clinicians and support and information for patients. THE MANUAL is not responsible for the content of this resource.
Leukemia & Lymphoma Society: Resources for Healthcare Professionals: provides educational resources for health care practitioners as well as information for patient referrals
Drug Information for the Topic





