



Cushing syndrome is a constellation of clinical manifestations caused by chronic high blood levels of cortisol or related glucocorticoids. Cushing disease is Cushing syndrome that results from excess pituitary production of adrenocorticotropic hormone (ACTH) secondary to a pituitary tumor. Typical symptoms and signs include moon face and truncal obesity, easy bruising, and thin arms and legs. Diagnosis is by history of receiving glucocorticoids or by finding elevated and/or relatively autonomous serum cortisol. Treatment depends on the cause.
(See also Overview of Adrenal Function.)
Etiology of Cushing Syndrome
Hyperfunction of the adrenal cortex can be dependent on adrenocorticotropic hormone (ACTH) or ACTH-independent.
ACTH-dependent hyperfunction may result from:
Hypersecretion of ACTH by the pituitary gland (Cushing disease)
Secretion of ACTH by a nonpituitary tumor, such as small cell carcinoma of the lung or a neuroendocrine tumor (ectopic ACTH syndrome)
Administration of exogenous ACTH
ACTH-independent hyperfunction usually results from:
Therapeutic administration of glucocorticoids
Adrenal adenomas or carcinomas
Rare causes of ACTH-independent hyperfunction include primary pigmented nodular adrenal dysplasia (usually in adolescents) and bilateral macronodular hyperplasia (in older adults).
Whereas the term Cushing syndrome denotes the clinical picture resulting from glucocorticoid excess from any cause, Cushing disease refers to hyperfunction of the adrenal cortex due to pituitary ACTH excess. Patients with Cushing disease almost always have a small tumor of the pituitary gland.
Symptoms and Signs of Cushing Syndrome
Clinical manifestations of Cushing syndrome include:
Facial rounding (called moon face) with a plethoric appearance
Truncal obesity with prominent supraclavicular and dorsal cervical fat pads (buffalo hump)
Striae (stretch marks)
Usually, very slender distal extremities and fingers


This photo shows characteristic facial rounding in a patient with Cushing syndrome.
This photo shows characteristic facial rounding in a patient with Cushing syndrome.
© Springer Science+Business Media

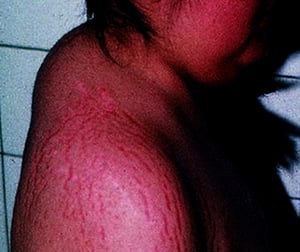
This patient with Cushing syndrome has characteristic buffalo hump and striae.
This patient with Cushing syndrome has characteristic buffalo hump and striae.
© Springer Science+Business Media

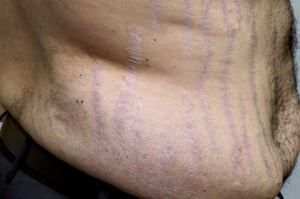
Linear stretch marks (striae) are visible on the abdomen of this patient with Cushing syndrome.
Linear stretch marks (striae) are visible on the abdomen of this patient with Cushing syndrome.
SCIENCE PHOTO LIBRARY

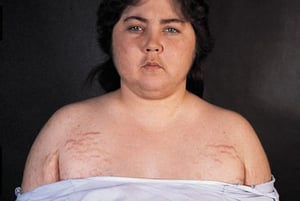
In this patient with Cushing syndrome, findings shown include facial rounding, plethora of the cheeks, supraclavicular fat, and striae.
In this patient with Cushing syndrome, findings shown include facial rounding, plethora of the cheeks, supraclavicular
By permission of the publisher. From Biller B. In Atlas of Clinical Endocrinology: Neuroendocrinology and Pituitary Disease. Edited by SG Korenman (series editor) and ME Molitch. Philadelphia, Current Medicine, 2000.
This photo shows characteristic facial rounding in a patient with Cushing syndrome.
This photo shows characteristic facial rounding in a patient with Cushing syndrome.
© Springer Science+Business Media
This patient with Cushing syndrome has characteristic buffalo hump and striae.
This patient with Cushing syndrome has characteristic buffalo hump and striae.
© Springer Science+Business Media
Linear stretch marks (striae) are visible on the abdomen of this patient with Cushing syndrome.
Linear stretch marks (striae) are visible on the abdomen of this patient with Cushing syndrome.
SCIENCE PHOTO LIBRARY
In this patient with Cushing syndrome, findings shown include facial rounding, plethora of the cheeks, supraclavicular fat, and striae.
In this patient with Cushing syndrome, findings shown include facial rounding, plethora of the cheeks, supraclavicular
By permission of the publisher. From Biller B. In Atlas of Clinical Endocrinology: Neuroendocrinology and Pituitary Disease. Edited by SG Korenman (series editor) and ME Molitch. Philadelphia, Current Medicine, 2000.
Muscle wasting and weakness are present. The skin is thin and atrophic, with poor wound healing and easy bruising. Striae may appear on the abdomen. Hypertension, renal calculi, osteoporosis, glucose intolerance, reduced resistance to infection, and mental disturbances are common. Cessation of linear growth is characteristic in children.
Females usually have menstrual irregularities. In females with adrenal tumors, increased production of androgens may lead to hirsutism, temporal balding, and other signs of virilism.
Diagnosis of Cushing Syndrome
Urinary free cortisol level
Dexamethasone suppression test
Midnight serum or salivary cortisol levels
Plasma ACTH levels; if detectable, provocative testing
Diagnosis is usually suspected based on the characteristic symptoms and signs. Confirmation (and identification of the cause) generally requires hormonal and imaging tests.
Urinary free cortisol measurement
Testing may begin with a 24-hour measurement of urinary free cortisol, which is elevated > 120 mcg/24 hours (> 331 nmol/24 hours) in almost all patients with Cushing syndrome. However, many patients with elevations of urinary free cortisol between 100 and 150 mcg/24 hours (276 and 414 nmol/24 hours) have obesity, depression, or polycystic ovaries but not Cushing syndrome. Normal ranges may vary according to assay.
A patient with suspected Cushing syndrome with grossly elevated urinary free cortisol (> 4 times the upper limit of normal) almost certainly has Cushing syndrome. Two to 3 normal collections usually exclude the diagnosis. Slightly elevated levels generally necessitate further investigation, as do normal levels when clinical suspicion is high.
A baseline morning (eg, 9 AM) serum cortisol measurement should also be done.
Dexamethasone suppression test
An alternative approach to investigation uses the dexamethasone suppression test, in which 1mg of dexamethasone is given orally at 11 to 12 PM and serum cortisol is measured at 8 to 9 AM the next morning. In most patients without Cushing syndrome, this medication suppresses morning serum cortisol to < 1.8 mcg/dL (< 50 nmol/L), whereas patients with Cushing syndrome virtually always have a higher level (1). A more specific but equally sensitive test is to give dexamethasone 0.5 mg orally every 6 hours for 2 days (low dose). In general, a clear failure to suppress cortisol levels in response to low-dose dexamethasone establishes the diagnosis, unless there is reason to suspect abnormal dexamethasone absorption or metabolism.
Previously, the high-dose dexamethasone suppression test (2 mg every 6hours for 48 hours) was used to differentiate Cushing disease (pituitary-dependent Cushing syndrome) from other causes of Cushing syndrome. Suppression of cortisol on high-dose testing suggests Cushing disease; lack of suppression suggests an adrenal gland tumor or ectopic ACTH secretion. However, most centers have abandoned its use because of its suboptimal sensitivity and specificity.
Midnight cortisol measurements
If results of urinary free cortisol measurements and the dexamethasone suppression test are indeterminate, the patient can be hospitalized for measurement of serum cortisol at midnight, which is more likely to be conclusive. Alternatively, and more conveniently, the patient may collect salivary cortisol samples and store them in the refrigerator at home. Serum cortisol normally ranges from 5 to 25 mcg/dL (138 to 690 nmol/L) in the early morning (6 to 8 AM) and declines gradually to < 1.8 mcg/dL (< 50 nmol/L) at midnight. Patients with Cushing syndrome occasionally have a normal morning serum cortisol level but lack normal diurnal decline in cortisol production, such that the midnight serum cortisol levels are above normal and the total 24-hour cortisol production may be elevated. Normal ranges of midnight salivary cortisol level vary according to assay, and it is advisable to collect several (2 to 3) samples. It may be that midnight salivary will prove to be more sensitive than measurement of midnight salivary cortisol (2).
Serum cortisol may be spuriously elevated in patients with congenital increases of glucocorticoid-binding globulin or in those receiving estrogen therapy, but diurnal variation is normal in these patients.
Plasma ACTH measurement
ACTH levels are measured to determine the cause of Cushing syndrome. Undetectable levels suggest a primary adrenal cause. High levels suggest a pituitary cause or an ectopic source. If ACTH is detectable, provocative tests help differentiate Cushing disease from ectopic ACTH syndrome, which is rarer.
In response to high-dose dexamethasone (2 mg orally every 6 hours for 48 hours), the 9 AM serum cortisol falls by > 50% in most patients with Cushing disease but only infrequently falls in those with ectopic ACTH syndrome, although most centers no longer use this test. Conversely, ACTH and cortisol rise in response to desmopressin (10 mcg IV, criteria vary) in most patients with Cushing disease but very rarely in those with ectopic ACTH syndrome (see table ). Corticotropin-releasing hormone (CRH) causes a similar rise in ACTH and cortisol and has been used in diagnostic testing; however, its availability is currently limited.
An alternative approach to localization, which is more accurate but more invasive, is to catheterize both petrosal veins (which drain the pituitary) and measure ACTH from these veins 5 minutes after giving a bolus of a 10-mcg IV dose of desmopressin. A central-to-peripheral ACTH ratio > 3 virtually excludes ectopic ACTH syndrome, whereas a ratio < 3 suggests a need to seek such a source.
Diagnostic Tests in Cushing Syndrome
Diagnosis | Serum Cortisol, 9 AM | Salivary or Serum Cortisol, Midnight | Urinary Free Cortisol | Low-Dose or Overnight Dexamethasone Suppression Test | High-Dose Dexamethasone Suppression Test | Desmopressin Stimulation Test | ACTH Level |
|---|---|---|---|---|---|---|---|
Normal | N | N | N* | S | S | Flat | N |
Cushing disease | N or ↑ | ↑ | ↑ | NS | S | ↑ | N or ↑ |
Ectopic ACTH | N or ↑ | ↑ | ↑ | NS | NS | Flat or ↑ (rarely) | ↑ or ↑↑ |
Adrenal tumor | N or ↑ | ↑ | ↑ | NS | NS | Flat | ↓ |
* May be elevated in non-Cushing conditions. | |||||||
ACTH = adrenocorticotropic hormone; Flat = no significant rise in ACTH or cortisol; N = normal; NS = nonsuppression; S = suppression; ↑↑= greatly increased; ↑= increased; ↓= decreased. | |||||||
Imaging
Pituitary imaging is done if ACTH levels and provocative tests suggest a pituitary cause; gadolinium-enhanced MRI is most accurate, but some microadenomas are visible on CT. If testing suggests a nonpituitary cause, imaging includes high-resolution CT of the chest, pancreas, and adrenals: scintiscanning or PET scanning with radiolabeled octreotide or, preferably, gallium-68 dotatate-PET, and occasionally fluorodeoxyglucose (FDG)-PET scanning. Petrosal sinus sampling may be needed to differentiate pituitary from ectopic sources. Several radionuclides specific for pituitary tumors are under development.
In children with Cushing disease, pituitary tumors are very small and usually cannot be detected with MRI. Petrosal sinus sampling is particularly useful in this situation. MRI is preferred to CT in pregnant patients to avoid fetal exposure to radiation.
Diagnosis references
1. Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847-875. doi:10.1016/S2213-8587(21)00235-7
2. Mohamed RS, Abuelgasim B, Barker S, et al. Late-night salivary cortisol and cortisone should be the initial screening test for Cushing's syndrome. Endocr Connect. 2022;11(7):e220050. doi:10.1530/EC-22-0050
Treatment of Cushing Syndrome
High protein intake and potassium administration (or potassium-sparing diuretics such as spironolactone)
Adrenal inhibitors such as metyrapone or ketoconazole and rarely mitotane, or medications such as osilodrostat and levoketoconazole
Surgery or radiation therapy to remove pituitary, adrenal, or ectopic ACTH-producing tumors
Sometimes somatostatin analogs or dopamine agonists to block ACTH secretion, or the glucocorticoid receptor antagonist mifepristone
Sometimes parenteral etomidate to inhibit 11-beta hydroxylase and reduce adrenal steroidogenesis
Initially, the patient’s general condition should be supported by high protein intake to counter protein catabolism and appropriate administration of potassium to treat hypokalemia. Hyperglycemia and hypertension should be treated in the usual manner, depending on severity, and it is generally now recommended to anticoagulate with prophylactic subcutaneous low molecular weight heparin.
If clinical manifestations of hypercortisolism are severe, it may be reasonable to block glucocorticoid secretion with metyrapone 250 mg to 1 g orally 3 times a day or ketoconazole 200-400 mg orally 2-3 times a day, increasing to a maximum of 400 mg 3 times a day. Ketoconazole is probably slower in onset and is sometimes hepatotoxic. Alternatives include mitotane, levoketoconazole, and osilodrostat, which block steroidogenesis, or mifepristone, which is a glucocorticoid receptor antagonist. Relacorilant, which is under development, acts as a glucocorticoid-receptor antagonist but, unlike mifepristone, does not affect the progesterone receptor. Parenteral etomidate (an intravenous anesthetic that also blocks cortisol production) may be life-saving for patients with fulminant symptoms; it is given as an intravenous infusion. The starting dose is usually 1 to 2 mg/hour, increasing as necessary, with frequent assessments of cortisol levels and dose titration accordingly.
ACTH-secreting pituitary tumors
Pituitary tumors that produce excessive ACTH are removed surgically or extirpated with radiation therapy. If no tumor is identified on imaging but a pituitary source is likely, total hypophysectomy may be attempted, particularly in older patients. Younger patients (including children and adolescents) may receive supervoltage irradiation of the pituitary, delivering 45 Gy (Gray). However, in children, irradiation may reduce secretion of growth hormone and occasionally cause precocious puberty. In special centers, a focused beam of radiation therapy may be given as a single dose (radiosurgery). Alternatively, proton beam therapy can be used if available, especially in children. Response to irradiation occasionally requires several years, but response is more rapid in children.
Studies suggest that mild cases of persistent or recurrent disease may benefit from medications that suppress ACTH secretion, including the somatostatin analog pasireotide and the dopamine agonist cabergoline. However, hyperglycemia is a significant adverse effect of pasireotide. Alternatively, the glucocorticoid receptors can be blocked with mifepristone. By blocking these receptors in the brain, mifepristone disrupts the negative feedback loop and increases serum cortisol, but it blocks the effects of the glucocorticoid and may cause hypokalemia. Because of these effects, biochemical monitoring is difficult.
Bilateral adrenalectomy is reserved for patients with pituitary hyperadrenocorticism who do not respond to both pituitary exploration (with possible adenomectomy) and irradiation, or in patients in whom surgery was unsuccessful and radiotherapy is contraindicated. Adrenalectomy requires life-long glucocorticoid replacement.
Glucocorticoid-secreting adrenocortical tumors
Adrenocortical tumors are removed surgically. Patients must receive cortisol during the surgical and postoperative periods because their nontumorous adrenal cortex will be atrophic and suppressed.
Benign adenomas can be removed laparoscopically.
With multinodular adrenal hyperplasia, bilateral adrenalectomy may be necessary, but in some cases removal of the larger adrenal alone may be effective. Even after a presumed total adrenalectomy, functional regrowth occurs in a few patients.
Ectopic ACTH-producing tumors
Ectopic ACTH syndrome is treated by removing the nonpituitary tumor that is producing the ACTH. However, in some cases, the tumor is disseminated and cannot be excised. Adrenal enzyme inhibitors, such as metyrapone 500 mg orally 3 times a day (and up to a total of 6 g a day) or the selective inhibitor of 11-beta-hydroxylase osilodrostat, usually control severe metabolic disturbances (eg, hypokalemia). Ketoconazole 400 to 1200 mg orally daily and levoketoconazole up to 1200 mg daily in divided doses also block glucocorticoid synthesis, although they may cause liver toxicity, QT interval prolongation leading to ventricular dysrhythmias, and addisonian symptoms (eg, weakness, fatigue, orthostatic hypotension, hyperpigmentation). It should be emphasized that medications blocking 11-beta-hydroxylase may lead to a build-up of precursors, which may falsely elevate ortisol levels in many assays, but not those based on GCMS (gas chromatography/mass spectrometry).
Mifepristone also may be useful for treating ectopic ACTH syndrome; however, because it blocks the action of cortisol but does not lower serum levels, monitoring its use can be difficult.
In an emergency situation, parenteral etomidate can produce a rapid fall in serum cortisol, but its use requires careful monitoring.
In the case of a disseminated ACTH-producing neuroendocrine tumor, bilateral adrenalectomy may be indicated.
Sometimes ectopic ACTH-secreting tumors respond to long-acting somatostatin analogs (eg, octreotide and/or others), although administration for > 2 years requires close follow-up because mild gastritis, gallstones, cholangitis, and malabsorption may develop.
Nelson syndrome
Nelson syndrome occurs when the pituitary gland continues to expand after bilateral adrenalectomy, causing a marked increase in the secretion of ACTH and its precursors and resulting in severe hyperpigmentation. It occurs in approximately 20 to 25% of patients who undergo adrenalectomy (1). The risk is probably reduced if the patient undergoes prophylactic pituitary radiation therapy at the time of adrenalectomy, but most centers simply scan the pituitary at frequent intervals with regular assessment of ACTH levels.
Although irradiation may arrest continued pituitary growth, many patients also require hypophysectomy. The indications for hypophysectomy are the same as for any pituitary tumor: an increase in size such that the tumor encroaches on surrounding structures, causing visual field defects, pressure on the hypothalamus, or other complications.
Radiation therapy may be given if it was not given at the time of bilateral adrenalectomy. Radiosurgery, or focused radiation therapy, can be given in a single fraction when standard external beam radiation therapy has already been done, as long as the lesion is at a reasonable distance from the optic nerve and chiasm.
Treatment reference
1. Reincke M, Albani A, Assie G, et al. Corticotroph tumor progression after bilateral adrenalectomy (Nelson's syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol. 2021;184(3):P1-P16. doi:10.1530/EJE-20-1088
Key Points
Diagnosis is usually made by elevated nocturnal serum or salivary cortisol levels, or 24-hour urinary free cortisol level, and a dexamethasone suppression test in which serum cortisol does not suppress.
Pituitary causes are distinguished from nonpituitary causes by adrenocorticotropic hormone (ACTH) levels.
Imaging is then done to identify any causative tumor.
Tumors are usually treated surgically or with radiation therapy.
Metyrapone, ketoconazole, levoketoconazole, or osilodrostat may be given to suppress adrenal cortisol secretion prior to definitive treatment or the glucocorticoid receptor antagonist mifepristone may be given. For urgent parenteral therapy, IV etomidate can be given with careful monitoring.
Pasireotide or cabergoline to suppress ACTH secretion may be given to patients with recurrent pituitary or disseminated ectopic ACTH-producing tumors.
Drug Information for the Topic





