



Gigantism and acromegaly are conditions caused by of excessive secretion of growth hormone (hypersomatotropism), nearly always due to a pituitary adenoma. Before closure of the epiphyses, the result is gigantism. Later, the result is acromegaly, which causes distinctive facial and other features. Diagnosis is clinical, by skull and hand radiographs, and by measurement of levels of growth hormone and insulin-like growth factor 1. Treatment involves removal or destruction of the responsible adenoma, and sometimes also other treatment modalities.
Growth hormone (GH) stimulates somatic growth and regulates metabolism. Growth hormone–releasing hormone (GHRH) is the major stimulator and somatostatin is the major inhibitor of the synthesis and release of GH. GH controls synthesis of insulin-like growth factor 1 (IGF-1, also called somatomedin-C), which largely controls growth. Although IGF-1 is produced by many tissues locally, the liver is the major source of circulating IGF-1. The metabolic effects of GH are biphasic. GH initially exerts insulin-like effects, increasing glucose uptake in muscle and fat, stimulating amino acid uptake and protein synthesis in liver and muscle, and inhibiting lipolysis in adipose tissue. Several hours later, more profound anti–insulin-like metabolic effects occur. They include inhibition of glucose uptake and use, causing blood glucose and lipolysis to increase, which increases plasma free fatty acids.
GH-secreting tumors are largely sporadic, but genetic abnormalities in the X chromosome (X-linked acrogigantism), overexpression of the pituitary tumor transforming gene (PTTG), and mutations in the aryl hydrocarbon receptor–interacting protein (AIP) have been documented. Some GH–secreting adenomas contain a mutant form of the Gs protein, which is a stimulatory regulator of adenylate cyclase. Cells with the mutant form of Gs protein secrete GH even in the absence of GHRH. A few cases of ectopic GHRH-producing tumors, especially of the pancreas and lung, also have been described.
Symptoms and Signs of Gigantism and Acromegaly
Pituitary gigantism


The patient on the right was diagnosed with gigantism as an infant. She is much taller than her mother on the left.
BETTINA CIRONE/SCIENCE PHOTO LIBRARY
This rare condition occurs if GH hypersecretion begins in childhood, before closure of the epiphyses. Skeletal growth velocity and ultimate stature are increased, but little bony deformity occurs. However, soft-tissue swelling occurs, and the peripheral nerves are enlarged. Delayed puberty or hypogonadotropic hypogonadism is also frequently present, resulting in a body build that is tall and slender with long extremities.
Acromegaly
In acromegaly, GH hypersecretion usually starts when the patient is between 20 and 40 years old. When GH hypersecretion begins after epiphyseal closure, the earliest clinical manifestations are coarsening of the facial features and soft-tissue swelling of the hands and feet. Appearance changes, and larger rings, gloves, and shoes are needed. Photographs of the patient are important in delineating the course of the disease.

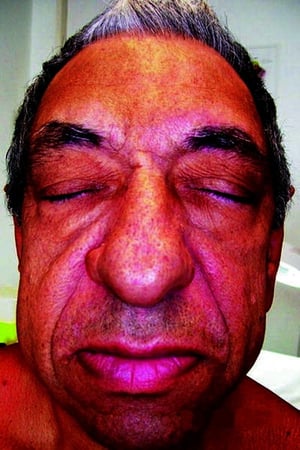
This photo shows a patient with frontal bossing, prognathism, nasal bone hypertrophy, and thickened skin.
This photo shows a patient with frontal bossing, prognathism, nasal bone hypertrophy, and thickened skin.
© Springer Science+Business Media


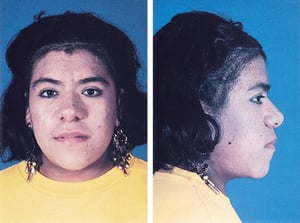
Left image shows a 64-year-old woman who presented with acromegaly due to a pituitary adenoma. Right image shows the same patient 11 years earlier. Note the change in facial features compared with the left image.
Left image shows a 64-year-old woman who presented with acromegaly due to a pituitary adenoma. Right image shows the sa
By permission of the publisher. From Newman C. In Atlas of Clinical Endocrinology: Neuroendocrinology and Pituitary Disease. Edited by SG Korenman (series editor) and ME Molitch. Philadelphia, Current Medicine, 2000.

Frontal and lateral images of a patient with acromegaly. Coarse facial features are evident, including prognathism and prominence of the malar eminences and supraorbital ridges.
Frontal and lateral images of a patient with acromegaly. Coarse facial features are evident, including prognathism and
By permission of the publisher. From Conrad C, Pro B, Prabhu S, et al. In Atlas of Cancer. Edited by M Markman and MR Gilbert. Philadelphia, Current Medicine, 2002.

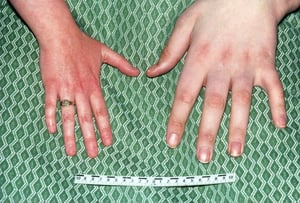
This photo shows a normal hand (on the left) compared to the enlarged hand (on the right) of a patient with acromegaly.
This photo shows a normal hand (on the left) compared to the enlarged hand (on the right) of a patient with acromegaly.
BIOPHOTO ASSOCIATES/SCIENCE PHOTO LIBRARY
This photo shows a patient with frontal bossing, prognathism, nasal bone hypertrophy, and thickened skin.
This photo shows a patient with frontal bossing, prognathism, nasal bone hypertrophy, and thickened skin.
© Springer Science+Business Media
Left image shows a 64-year-old woman who presented with acromegaly due to a pituitary adenoma. Right image shows the same patient 11 years earlier. Note the change in facial features compared with the left image.
Left image shows a 64-year-old woman who presented with acromegaly due to a pituitary adenoma. Right image shows the sa
By permission of the publisher. From Newman C. In Atlas of Clinical Endocrinology: Neuroendocrinology and Pituitary Disease. Edited by SG Korenman (series editor) and ME Molitch. Philadelphia, Current Medicine, 2000.
Frontal and lateral images of a patient with acromegaly. Coarse facial features are evident, including prognathism and prominence of the malar eminences and supraorbital ridges.
Frontal and lateral images of a patient with acromegaly. Coarse facial features are evident, including prognathism and
By permission of the publisher. From Conrad C, Pro B, Prabhu S, et al. In Atlas of Cancer. Edited by M Markman and MR Gilbert. Philadelphia, Current Medicine, 2002.
This photo shows a normal hand (on the left) compared to the enlarged hand (on the right) of a patient with acromegaly.
This photo shows a normal hand (on the left) compared to the enlarged hand (on the right) of a patient with acromegaly.
BIOPHOTO ASSOCIATES/SCIENCE PHOTO LIBRARY
In adults with acromegaly, coarse body hair increases, and the skin thickens and frequently darkens. The size and function of sebaceous and sweat glands increase, such that patients frequently complain of excessive perspiration and offensive body odor. Overgrowth of the mandible leads to protrusion of the jaw (prognathism) and malocclusion of teeth. Cartilaginous proliferation of the larynx leads to a deep, husky voice. The tongue is frequently enlarged and furrowed. In long-standing acromegaly, costal cartilage growth leads to a barrel chest. Articular cartilaginous proliferation occurs early in response to GH excess, with the articular cartilage possibly undergoing necrosis and erosion. Joint symptoms are common, and crippling degenerative arthritis may occur.
Peripheral neuropathies occur commonly because of compression of nerves by adjacent fibrous tissue and endoneural fibrous proliferation. Headaches are common because of the pituitary tumor. Bitemporal hemianopia may develop if suprasellar extension compresses the optic chiasm. The heart, liver, kidneys, spleen, thyroid gland, parathyroid glands, colon, and pancreas are larger than normal; thyroid enlargement may be generalized or multinodular.
Cardiac disease (eg, coronary artery disease, cardiomegaly, valvular insufficiencies, sometimes cardiomyopathy) occurs in perhaps one-third of patients, with a doubling in the risk of death due to cardiac disease. Hypertension occurs in up to one-third of patients.
Snoring is a common symptom, and obstructive sleep apnea occurs in 40 to 50% of patients.
Colonic polyps are increased as a result of GH excess. The risk of cancer, particularly of the gastrointestinal tract, increases 2-fold to 3-fold. GH increases tubular reabsorption of phosphate and leads to mild hyperphosphatemia.
Impaired glucose tolerance occurs in nearly half the patients with acromegaly and in gigantism, but clinically significant diabetes mellitus occurs in only approximately 10% of patients.
Galactorrhea occurs in some women with acromegaly, usually in association with hyperprolactinemia. However, galactorrhea may occur with GH excess alone, because GH itself stimulates lactation. Decreased gonadotropin secretion often occurs with GH-secreting tumors. Approximately one-third of males with acromegaly develop erectile dysfunction, and nearly all females develop menstrual irregularities or amenorrhea.
Diagnosis of Gigantism and Acromegaly
CT or MRI
Insulin-like growth factor 1 (IGF-1) levels
Usually growth hormone (GH) levels
Diagnosis can be made from the characteristic clinical findings in the setting of characteristic hormone abnormalities (1, 2). MRI of the sella is the imaging test of choice for diagnosis of pituitary adenoma. CT, MRI, or skull radiographs disclose cortical thickening, enlargement of the frontal sinuses, and enlargement and erosion of the sella turcica. Radiographs of the hands show tufting of the terminal phalanges and soft-tissue thickening.
Serum IGF-1 should be measured in patients with suspected acromegaly; IGF-1 levels are typically substantially elevated (3-fold to 10-fold), and because IGF-1 levels do not fluctuate like GH levels do, they are the simplest way to assess GH hypersecretion. IGF-1 levels also can be used to monitor response to therapy.
Plasma GH levels are typically elevated. Blood should be taken before the patient eats breakfast (basal state); in normal people, basal GH levels are low or undetectable. Transient elevations of GH are normal, due to the pulsatile secretion of GH, and must be distinguished from pathologic hypersecretion. The degree of GH suppression after a glucose load remains the standard and thus should be measured in patients with elevated plasma GH; however, the results are assay-dependent, and the cutoff for normal suppression is controversial. Secretion in normal people is suppressed to < 1 ng/mL ([< 1 mcg/L]; a cutoff of < 0.4 ng/mL [< 0.4 mcg/L] is often used) within 120 minutes of oral administration of 75 g of glucose. Most patients with acromegaly have substantially higher values. In some cases, basal plasma GH levels are also used in monitoring response to therapy.
CT or MRI of the sella should be done to evaluate for a tumor. If a tumor is not visible, excessive secretion of pituitary GH may be due to a tumor outside the central nervous system producing excessive amounts of ectopic GHRH. Demonstration of elevated levels of plasma GHRH can confirm the diagnosis. Lungs and pancreas may be first evaluated in searching for the sites of ectopic production.
Screening for complications
Screening for complications, including diabetes, heart disease, and gastrointestinal cancer, should be done at the time of diagnosis.
Fasting plasma glucose levels, glycosylated Hb (HbA1C), or an oral glucose tolerance test can be done to test for diabetes.
Electrocardiography and, preferably, echocardiography are done to detect heart disease.
Colonoscopy is done to detect colon polyps and cancer.
Follow-up screening depends on the results of the initial testing and the patient's response to treatment.
Diagnosis references
1. Giustina A, Biermasz N, Casanueva FF, et al. Consensus on criteria for acromegaly diagnosis and remission [published correction appears in Pituitary. 2024 Feb;27(1):88. doi: 10.1007/s11102-023-01373-w]. Pituitary. 2024;27(1):7-22. doi:10.1007/s11102-023-01360-1
2. Cardosos LM, Marques P, Pereira MT, et al. Diagnosis and management of acromegaly: a consensus statement of the Pituitary Study Group of the Portuguese Society of Endocrinology, Diabetes and Metabolism. Endocrinol Insights. 2025;20:29-58. doi.org/10.1159/000541671
Treatment of Gigantism and Acromegaly
Surgery or radiation therapy
Sometimes pharmacologic suppression of GH secretion or activity
Surgical therapy
Selective removal of the pituitary tumor with surgery is considered first-line therapy for most patients (1). Patients with comorbidities that prevent safe surgical resection and those with unresectable tumors may be treated with primary medical therapy. Rates of remission after surgical resection are dependent on the size and degree of invasion of the pituitary adenoma and the experience of the neurosurgeon.
Surgical removal of the tumor is likely to have been curative if GH levels measured after a glucose load and IGF-1 levels reach normal values. If one or both values are abnormal, further therapy is usually needed. If GH excess is poorly controlled, hypertension, heart failure, and increased mortality occur.
Pharmacotherapy
In general, pharmacotherapy is indicated:
If surgery is contraindicated
If surgery or radiation therapy has not been curative
If radiation therapy is being given time to work
Medications available for the treatment of acromegaly include those that target tumor secretion of GH and one that blocks GH at the level of the GH receptor.
Somatostatin receptor ligands are a mainstay of therapy because they decrease GH secretion from the pituitary tumor mediated through interactions predominately with the somatostatin subtype receptor-2 (SSTR-2). Medications in this class include octreotide and lanreotide, which have high affinity for SSTR-2 and come in short-acting (octreotide) and long-acting (octreotide and lanreotide) preparations. Pasireotide, a somatostatin receptor ligand with affinity for SSTR-1, 2, 3, and 5, is also available in short- and long-acting preparations. All somatostatin receptor ligands may also cause tumor shrinkage.
Octreotide is started with an intramuscular injection delivered monthly and titrated up or down if needed to an effective dose after the third injection. Effective doses range from 10 to 40 mg monthly. Octreotide is also available in an oral preparation that is given twice daily.
Lanreotide is given as a deep subcutaneous injection monthly, with the possibility of extended dosing ( every 6 to 8 weeks) in patients with well-controlled disease.
Pasireotide is generally considered if octreotide or lanreotide is unsuccessful in returning IGF-I levels to normal.
Cabergoline, a dopamine agonist, has been used alone or in combination with a somatostatin receptor ligand and works by suppressing GH secretion at the pituitary. Cabergoline is typically used in mild disease and has the advantage of being an oral medication.
Pegvisomant, the GH receptor antagonist, is given as a daily subcutaneous injection and decreases IGF-I levels and symptoms but does not decrease GH levels or act on the pituitary tumor. Pegvisomant is titrated to an effective dose depending on IGF-I levels.
Radiation therapy
Radiation therapy may be used at any step of treatment but is typically used as primary therapy only when surgery or medications are not available. Timing of radiation in the treatment of patients with acromegaly varies among institutions. Stereotactic radiation, delivering approximately 5000 cGy to the pituitary, is used, but GH levels may not fall to normal for several years. Treatment with accelerated protons (heavy particle radiation) permits delivery of larger doses of radiation (equivalent to 10,000 cGy) to the pituitary; such therapy poses higher risk of cranial nerve and hypothalamic damage and is available only in a few centers.
Hypopituitarism commonly develops several years after irradiation. Because radiation damage is cumulative, proton beam therapy should not be used after conventional gamma-irradiation. A combined approach with both surgery and radiation therapy is indicated for patients with progressive extrasellar involvement by a pituitary tumor and for patients in whom the entire tumor cannot be resected, which is often the case.
Treatment reference
1. Giustina A, Biermasz N, Casanueva FF, et al. Consensus on criteria for acromegaly diagnosis and remission [published correction appears in Pituitary. 2024 Feb;27(1):88. doi: 10.1007/s11102-023-01373-w]. Pituitary. 2024;27(1):7-22. doi:10.1007/s11102-023-01360-1
Prognosis of Gigantism and Acromegaly
Predictors of mortality in acromegaly include hypertension, age, use of radiation, and hypopituitarism, especially adrenocorticotropic hormone deficiency. Reduction of IGF-I and GH levels to the normal range seems to reduce mortality rates to normal.
Key Points
Gigantism and acromegaly are usually caused by a pituitary adenoma that secretes excessive amounts of growth hormone (GH); rarely, they are caused by non-pituitary tumors that secrete growth hormone–releasing hormone (GHRH).
Gigantism occurs if GH hypersecretion begins in childhood, before closure of the epiphyses.
Acromegaly involves GH hypersecretion beginning in adulthood; a variety of bony and soft tissue abnormalities develop.
Diagnose by measuring insulin-like growth factor 1 (IGF-1) and GH levels; do central nervous system imaging to detect a pituitary tumor.
Remove pituitary tumors surgically or using radiation therapy.
If tumors cannot be removed, give octreotide or lanreotide to suppress GH secretion, or pegvisomant to block GH-induced IGF-1 secretion.
Drug Information for the Topic





