



The immune system consists of cellular components and molecular components that work together to destroy antigens. (See also Overview of the Immune System.)
Antigen-Presenting Cells
Although some antigens (Ags) can stimulate the immune response directly, T cell–dependent acquired immune responses typically require antigen-presenting cells (APCs) to present antigen-derived peptides within major histocompatibility complex (MHC) molecules (1).
Intracellular antigens (eg, viruses) can be processed and presented to CD8 cytotoxic T cells by any nucleated cell because all nucleated cells express class I MHC molecules. By encoding proteins that interfere with this process, some viruses (eg, cytomegalovirus) can evade elimination.
Extracellular antigens (eg, from many bacteria) that are phagocytosed or endocytosed can be processed into peptides and complexed with surface class II MHC molecules on professional APCs, which specialize in presenting antigens to CD4 helper T (Th) cells. The following cells constitutively express class II MHC molecules and therefore act as professional APCs:
Dendritic cells
Monocytes
Macrophages
Dendritic cells are present in the skin (as Langerhans cells), lymph nodes, and tissues throughout the body. (2) Dendritic cells in the skin act as sentinel APCs, taking up antigen, then traveling to local lymph nodes where they can activate T cells.
Plasmacytoid dendritic cells are a distinct type of dendritic cell, but are related to conventional dendritic cells. They are capable of acting as antigen-presenting cells, but are particularly specialized to produce large amounts of interferon-alpha and interferon-beta (3).
Follicular dendritic cells are another distinct lineage (namely, a different cell type than conventional dendritic cells) that do not express class II MHC molecules, and therefore do not present antigen to Th cells. They are not phagocytic; they have receptors for the crystallizable fragment (Fc) region of immunoglobulin (Ig) G and for complement, which enable them to bind to immune complexes and present the complex to B cells in germinal centers of secondary lymphoid organs (4).
Monocytes in the circulation are precursors to tissue macrophages. Monocytes migrate into tissues, where they rapidly develop into macrophages. This development occurs under the influence of macrophage colony-stimulating factor (M-CSF), which is secreted by various cell types (eg, endothelial cells, fibroblasts). At infection sites, activated T cells secrete cytokines (eg, interferon-gamma [IFN-gamma]) that induce the production of macrophage migration inhibitory factor, preventing macrophages from leaving.
Macrophages are phagocytic cells present in tissues throughout the body. Depending upon the activation signals they receive, macrophages can alter their gene expression profiles and differentiate into M1 or M2 subsets (see table ). M1, "classically activated," pro-inflammatory macrophages are stimulated by cytokines such as IFN-gamma and by various microbial components (eg, lipopolysaccharide from Gram-negative bacteria). M2 "alternatively activated" anti-inflammatory macrophages are stimulated predominantly by cytokines such as interleukin-4 (IL-4) and IL-13. M1 macrophages are strongly microbicidal, promote Th1 responses, and secrete proinflammatory cytokines (eg, tumor necrosis factor-alpha [TNF-alpha]), whereas M2 macrophages secrete immunosuppressive cytokines (eg, IL-10, transforming growth factor-beta [TGF-beta]), are important in resolving inflammation and in promoting tissue remodelling and function analogously to T regulatory cells. M2 macrophages can also contribute to fibrosis by producing profibrotic factors such as TGF-beta.
B cells' primary function is to develop into plasma cells, which manufacture and secrete antibodies.
Macrophage Subtypes
Characteristics | M1 | M2 |
|---|---|---|
Examples of activation agent | Lipopolysaccharide IFN-gamma (a cytokine produced by Th1 cells) | IL-4 and IL-13 (cytokines produced by Th2 cells) |
Examples of cytokines produced | Proinflammatory cytokines (eg, TNF-alpha) | Immunosuppressive cytokines (eg, IL-10, TGF-beta) |
Other functions | Promote Th1 responses Strongly microbicidal | Resolve inflammation Promote tissue remodelling |
IFN = interferon; IL = interleukin; TGF = transforming growth factor; Th1 cells = type 1 helper T cells; Th2 cells = type 2 helper T cells; TNF = tumor necrosis factor. | ||
Antigen-presenting cells references
1. Hoelting TLB, Park T, Brown CC. Antigen-presenting cells as arbiters of mucosal tolerance and immunity. Nat Immunol. 2025;26(11):1890-1902. doi:10.1038/s41590-025-02320-6
2. Shortman K. Dendritic cell development: A personal historical perspective. Mol Immunol. 2020;119:64-68. doi:10.1016/j.molimm.2019.12.016
3. Adams NM, Das A, Yun TJ, Reizis B. Ontogeny and Function of Plasmacytoid Dendritic Cells. Annu Rev Immunol. 2024;42(1):347-373. doi:10.1146/annurev-immunol-090122-041105
4. Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. 2014;14(7):495-504. doi:10.1038/nri3689


Monocyte
Eosinophil
Neutrophil
Basophil
Red blood cell (erythrocyte)
Lymphocyte
KATERYNA KON/SCIENCE PHOTO LIBRARY
Lymphocytes
The 2 main types of lymphocytes are:
B cells, which mature in the bone marrow
T cells, which mature in the thymus
The main types of lymphocytes are morphologically indistinguishable but have different immune functions. They can be distinguished by antigen-specific surface receptors and other cell surface molecules called clusters of differentiation (CDs), whose presence (+) or absence (-) define some subsets. Hundreds of CDs have been identified many of which are absent from lymphocytes but present on other cells of the immune system (1). CD molecules function in cell adhesion, cell signaling, as receptors for the Fc region of immunoglobulins, as receptors for components of the complement system, and others. (For further information on CD molecules, see the Human Cell Differentiation Molecules web site.) Each lymphocyte recognizes a specific antigen via cell surface B-cell receptors (BCRs, transmembrane antibody) or T-cell receptors (TCRs).
B cells
Approximately 5 to 10% of lymphocytes in the blood are B cells (2); they are also present in the bone marrow, spleen, lymph nodes, mucosa-associated lymphoid tissues, and to a lesser extent in non-lymphoid tissues (3).
B cells can present antigen to T cells and release cytokines, but their primary function is to develop into plasma cells, which manufacture and secrete antibodies.
Patients with B-cell immunodeficiencies (eg, X-linked agammaglobulinemia) are especially susceptible to recurrent bacterial infections (4, 5).
Through random rearrangement of the immunoglobulin's (Ig) variable (V), diversity (D), and joining (J) gene segments that encode the region of an antibody that is variable, B cells collectively have the potential to recognize an almost limitless number of unique antigens (6, 7, 8). Gene rearrangement occurs in programmed steps in the bone marrow during B-cell development. The process starts with a committed stem cell, continues through pro‒B cell and pre‒B cell stages, and results in an immature B cell (9, 10). At this point, any cells that interact with self antigen (autoimmune cells) are removed from the immature B cell population via inactivation (anergy) or apoptosis, thereby ensuring immune tolerance. Cells that are not removed (ie, those that recognize nonself antigen) continue to develop into mature naive B cells, leave the bone marrow, and enter peripheral lymphoid organs, where they may encounter antigens (11).
The mature naive B-cell response to antigen has 2 stages:
Primary immune response: When mature naive B cells first encounter an antigen, they become lymphoblasts, undergo clonal proliferation, and differentiate into memory cells, which have the ability to respond to the same antigen in the future, or into mature antibody-secreting plasma cells. After first exposure, there is a latent period of days before any antibody is produced. Initially, only IgM is produced. After that initial period, with the help of T cells, B cells can further rearrange their Ig genes and undergo class switching to produce antigen-specific IgG, IgA, or IgE. Thus, at first exposure, the antibody response is slow and initially provides limited protective immunity.
Secondary (anamnestic or booster) immune response: When memory B cells and Th cells are re-exposed to the same antigen, the memory B cells rapidly proliferate, differentiate into mature plasma cells, and promptly produce large amounts of antibody (chiefly IgG because of a T cell–induced class switch). The resulting antibody also has stronger binding affinity to the antigen due to mutation in the genes encoding the antibody variable regions. The antibody is then released into the blood and other tissues, where it can react with antigen. Thus, after re-exposure, the immune response is typically faster and more effective.
T cells
T cells develop from bone marrow stem cells that travel to the thymus, where they go through a process of rigorous selection (12). There are 3 main types of T cells:
Helper
Regulatory (suppressor)
Cytotoxic
In thymic selection, T cells that react to self antigen presented by self MHC molecules (or react strongly to self MHC molecules regardless of the antigen presented) are eliminated by apoptosis, limiting the likelihood of autoimmunity. Only T cells that can recognize nonself antigen complexed to self MHC molecules survive; they leave the thymus for the peripheral blood and lymphoid tissues.
Most mature T cells express either CD4 or CD8 and have an antigen-binding, Ig-like, surface receptor called the T-cell receptor (TCR). There are 2 types of TCR:
Alpha-beta TCR: Composed of TCR alpha and beta chains; present on most T cells
Gamma-delta TCR: Composed of TCR gamma and delta chains; present on a minor population of T cells
Genes that encode the TCR, similar to Ig genes, are rearranged, resulting in defined specificity and affinity for antigen (13). Most T cells (those with an alpha-beta TCR) recognize antigen-derived peptides displayed along with the MHC molecule of an antigen-presenting cell. T cells with gamma-delta TCR can recognize protein antigens directly or recognize lipid antigens displayed by an MHC-like molecule called CD1. Consequently, the number of T-cell specificities is almost limitless.
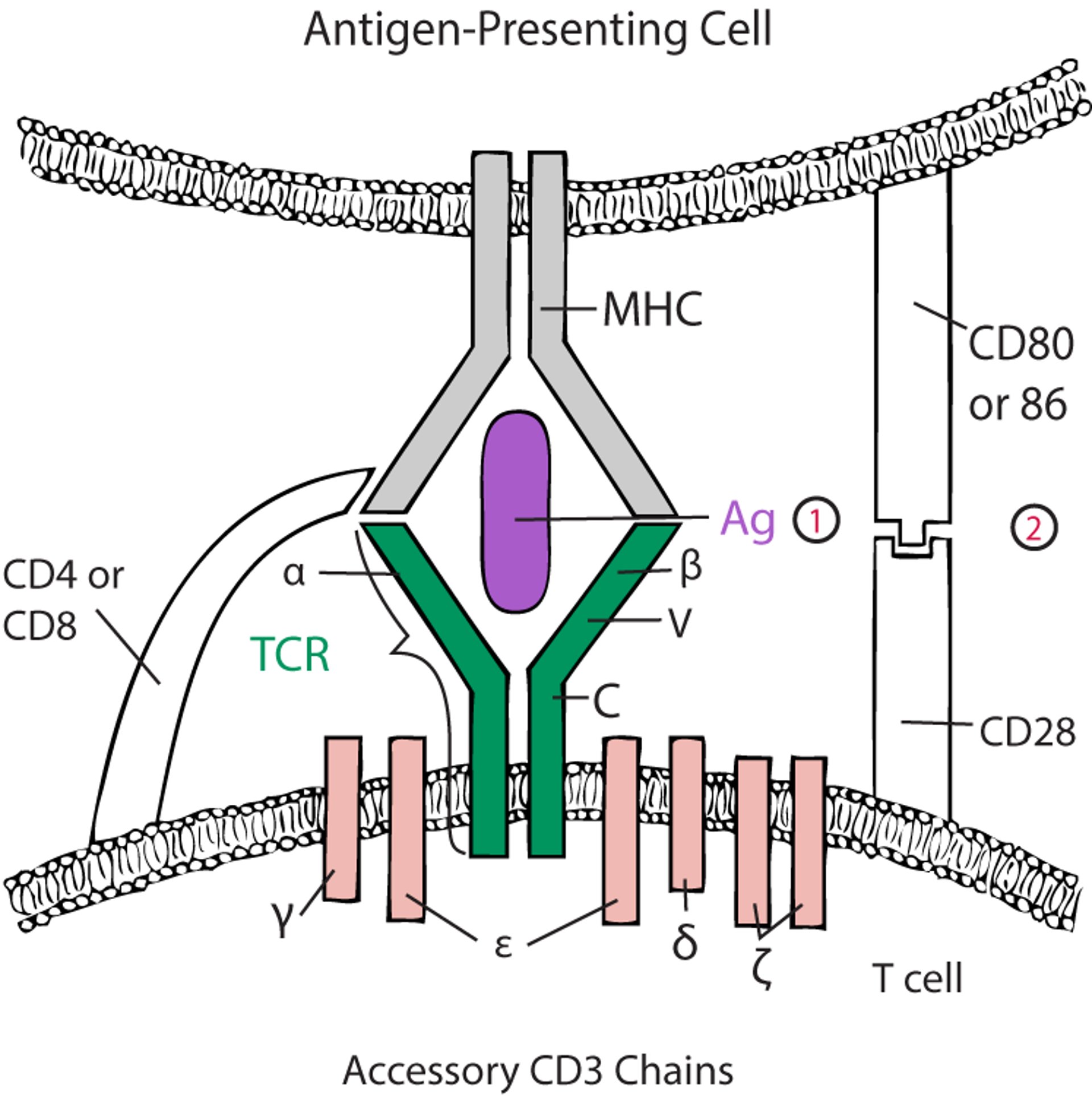
For alpha-beta T cells to be activated, the TCR must engage with antigen-MHC (see figure ). Costimulatory accessory molecules must also interact (eg, CD28 on the T cell interacts with CD80 and CD86 on the antigen-presenting cell); otherwise, the T cell exposed to antigen becomes anergic or dies by apoptosis (14, 15). Some accessory molecules (eg, CTLA-4 [cytotoxic T-lymphocyte antigen 4] and PD-1 [programmed cell death protein 1]) on the T cell, interact with ligands on antigen-presenting cells (CD80/CD86 and PD-L1, respectively) to inhibit previously activated T cells and thus dampen the immune response.
Molecules such as CTLA-4 and PD-1 and their ligands, are termed checkpoint molecules because they denote that the T cell needs to be restrained from continuing its activity (16). Cancer cells that express checkpoint molecules may thus be protected from the immune system by restraining the activity of tumor-specific T cells.
Monoclonal antibodies that target checkpoint molecules on either T cells or on tumor cells (termed checkpoint inhibitors, see table ) are used to prevent downregulation of antitumor immune responses and effectively treat some cancers and boost the antitumor response. However, because checkpoint molecules are also involved in preventing other types of immune response (such as self-directed autoimmune reactions), checkpoint inhibitors can permit severe immune-related inflammatory and autoimmune reactions to occur (both systemic and organ specific) or exacerbate autoimmune disorders (17, 18).
Polymorphisms in the CTLA-4 gene are associated with certain autoimmune disorders, including multiple sclerosis, Graves disease, and type 1 diabetes mellitus.
Two-Signal Model for T -Cell Activation
The alpha (α) and beta (β) chains of the T-cell receptor (TCR) bind to antigen (Ag)–major histocompatibility complex (MHC) on an antigen-presenting cell (APC), and CD4 or CD8 interacts with the MHC. Both interactions stimulate the T cell (first signal) through the accessory CD3 chains. However, without coactivation (second signal), the T cell is deemed anergic or tolerant. The TCR is largely structurally homologous to the B-cell receptor; the α and β (or gamma [γ] and delta [δ]) chains have constant (C) and variable (V) regions. (1) = first signal; (2) = second signal. |

Helper T (Th) cells primarily express CD4+ but may rarely express CD8+. They differentiate from Th0 (undifferentiated T helper) cells into one of the following subtypes:
Th1 cells: In general, Th1 cells promote cell-mediated immunity via cytotoxic T cells and macrophages and are thus particularly involved in defense against intracellular pathogens (eg, viruses) or cancer cells. They can also promote the production of some antibody classes (eg, subclasses of IgG such as IgG2 and IgG3).
Th2 cells: Th2 cells are particularly adept at promoting antibody production by B cells (humoral immunity) and are thus particularly involved in directing responses aimed at extracellular pathogens (eg, bacteria, fungi, parasites); they are also responsible for allergic inflammation and the production of IgE (19).
Th9 cells: Th9 cells promote inflammation and are involved in the regulation of immune responses.
Th17 cells: Th17 cells can promote tissue inflammation, and similar to Th2 cells, promote immunity to extracellular pathogens (eg, bacteria, fungi).
Th22 cells: Th22 cells protect epithelial barriers (20).
Tfh cells (T follicular helper cells): Tfh cells promote B-cell responses in the germinal centers of secondary lymphoid tissues (21).
Each cell type secretes several cytokines (see table ). Different patterns of cytokine production identify other Th-cell functional phenotypes. Depending on the stimulating pathogen, Th1 and Th2 cells can, to a certain extent, downregulate each other's activity, leading to dominance of a Th1 or a Th2 response.
Any T cells that can recognize peptides derived from self antigens are normally eliminated during T cell development in the thymus. If such T cells are not deleted, they can potentially develop into autoimmune Th1, Th2, Th9, Th17, or Th22 cells that may drive the development of autoimmune disease.
Functions of T Cells
Type | Characteristic Transcription Factor | Substances Produced | Primary Function |
|---|---|---|---|
Th1 | T-bet | IFN-gamma IL-2 Lymphotoxin (TNF-beta) TNF (TNF-alpha) | Facilitate macrophage and cytotoxic T-cell responses Fighting intracellular infections and tumors |
Th2 | GATA3 | IL-4 IL-5 IL-10 IL-13 | Stimulate antibody production by B cells Promoting allergic inflammation Fighting parasitic infestation |
Th9 | PU.1 | IL-9 | Promote inflammatory responses |
Th17 | RORgammaT | IL-17 IL-21 IL-22 | Promote inflammatory responses |
Th22 | AhR | IL-10 IL-13 IL-21 IL-22 | Promote epithelial barrier integrity by facilitating tissue repair and wound healing |
Tfh | Bcl-6 | IL-4 IL-10 IL-21 | Help germinal center B cells |
Tc | Subsets of Tc cells (Tc1, Tc2, Tc9, Tc17, and Tc22) are analogous to Th subsets in their characteristic transcription factors | Perforin Granzymes FasL Cytokines* | Kill infected cells |
Regulatory | Foxp3 | TGF-beta IL-10 IL-35 | Suppress immune responses |
NKT cells | Subsets of NKT cells have been described with characteristic transcription factors and cytokine production similar to Th and Tc subsets | Cytokines* | May help regulate immune responses |
* Tc, also known as CTL, can be subdivided into Tc1, Tc2, Tc9, Tc17, and Tc22 based upon analogous cytokine production to Th1, Th2, Th9, Th17, and Th22 cells. | |||
FasL = Fas ligand; IFN = interferon; IL = interleukin; NK = natural killer; Tc = cytotoxic T cell; TGF = transforming growth factor; Th = helper T cell; TNF = tumor necrosis factor. | |||
The distinction between the different Th cells is clinically relevant (22). For example, a Th1 response dominates in tuberculoid leprosy, and a Th2 response dominates in lepromatous leprosy. A Th1 response is characteristic of certain autoimmune disorders (eg, type 1 diabetes mellitus, multiple sclerosis). A Th2 response promotes IgE production and the development of allergic disorders, as well as helps B cells produce autoantibodies in some autoimmune disorders (eg, Graves disease, myasthenia gravis). Th17 cells, via their role in inflammation, may also contribute to autoimmune disorders such as psoriasis (23) and rheumatoid arthritis (24). Patients with immunodeficiencies characterized by defective Th17 cells (eg, hyper-IgE [Job] syndrome) are especially susceptible to infection with Candida albicans and Staphylococcus aureus.
Regulatory (suppressor) T (Treg) cells mediate suppression of immune responses and usually express the Foxp3 transcription factor (25). They comprise functional subsets of CD4+ or CD8+ expressing T cells that also express Foxp3+ :
Natural Treg cells develop within the thymus.
Induced Treg cells develop from conventional T cells upon encounter with antigen in the periphery.
Regulatory T cells secrete cytokines with immunosuppressive properties (eg, transforming growth factor [TGF]-beta and interleukin [IL]-10) deplete immunostimulatory interleukin-2 (IL-2) by expressing high levels of the CD25 component of the IL-2 receptor, or suppress the immune response by mechanisms that require cell-to-cell contact and involve cell surface molecules (eg, CTLA-4). The main role of Treg cells is to prevent excessive immune responses by contributing to the resolution of inflammation and by curbing allergic and autoimmune activity (26). Patients with functional mutations in FOXP3 develop the autoimmune disorder IPEX syndrome (immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome).
Cytotoxic T (Tc, also referred to as CTL) cells are primarily CD8+ expressing T cells. A small subset of CD4+ T cells can acquire cytotoxic function (27). They are vital for eliminating intracellular pathogens, especially viruses. Tc cells play a role in organ transplant rejection.
CTL development involves 3 phases:
A precursor cell that, when appropriately stimulated, can differentiate into a CTL
An effector cell that has differentiated and can kill its appropriate target
A memory cell that is quiescent (no longer stimulated) but is ready to become an effector when restimulated by the original antigen-MHC combination
Fully activated CTLs, like natural killer (NK) cells, can kill an infected target cell by inducing apoptosis.
CTL (Tc) can secrete cytokines and, like Th cells, have been divided into types Tc1, Tc2, Tc9, Tc17, and Tc22 based on their patterns of cytokine production (28).
TCL may be:
Syngeneic: Generated in response to self (autologous) cells whose MHC presents peptides derived from viral infection or other foreign proteins
Allogeneic: Generated in response to cells that express foreign MHC products (eg, in organ transplantation when the donor’s MHC molecules differ from the recipient’s)
Some Tc cells can directly recognize foreign MHC (direct pathway); others may recognize fragments of foreign MHC presented by self MHC molecules of the transplant recipient (indirect pathway).
Natural killer T (NKT) cells are a distinct subset of T cells that express CD56+ and CD16+ (29) as well as the pan T cell marker CD3+. Activated NKT cells may help regulate immune responses. NKT cells differ from NK cells in phenotype and certain functions.
Lymphocytes references
1. Fernández-Calles J, Kužílková D, Hedin F, et al. CD Molecules Nomenclature 2025: Antibody Validation and Expression Profiling of Immune System G Protein-Coupled Receptors. Eur J Immunol. 2025;55(12):e70099. doi:10.1002/eji.70099
2. Stock W, Hoffman R. White blood cells 1: non-malignant disorders. Lancet. 2000;355(9212):1351-1357. doi:10.1016/S0140-6736(00)02125-5
3. Samiea A, Celis G, Yadav R, Rodda LB, Moreau JM. B cells in non-lymphoid tissues. Nat Rev Immunol. 2025;25(7):483-496. doi:10.1038/s41577-025-01137-6
4. Smith T, Cunningham-Rundles C. Primary B-cell immunodeficiencies. Hum Immunol. 2019;80(6):351-362. doi:10.1016/j.humimm.2018.10.015
5. Tangye SG, Nguyen T, Deenick EK, Bryant VL, Ma CS. Inborn errors of human B cell development, differentiation, and function. J Exp Med. 2023;220(7):e20221105. doi:10.1084/jem.20221105
6. Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology. 2020;160(3):233-247. doi:10.1111/imm.13176
7. Jung D, Alt FW. Unraveling V(D)J recombination; insights into gene regulation. Cell. 2004;116(2):299-311. doi:10.1016/s0092-8674(04)00039-x
8. Sun A, Novobrantseva TI, Coffre M, et al. VH replacement in primary immunoglobulin repertoire diversification. Proc Natl Acad Sci U S A. 2015;112(5):E458-E466. doi:10.1073/pnas.1418001112
9. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570-1580. doi:10.1182/blood-2008-02-078071
10. Melchers F. Checkpoints that control B cell development. J Clin Invest. 2015;125(6):2203-2210. doi:10.1172/JCI78083
11. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell. 2019;177(3):524-540. doi:10.1016/j.cell.2019.03.016
12. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function throughout Life. Immunity. 2018;48(2):202-213. doi:10.1016/j.immuni.2018.01.007
13. Krangel MS. Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol. 2009;21(2):133-139. doi:10.1016/j.coi.2009.03.009
14. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227-242. doi:10.1038/nri3405
15. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 Costimulation: From Mechanism to Therapy. Immunity. 2016;44(5):973-988. doi:10.1016/j.immuni.2016.04.020
16. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106. doi:10.1097/COC.0000000000000239
17. Ibis B, Aliazis K, Cao C, Yenyuwadee S, Boussiotis VA. Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases. Front Immunol. 2023;14:1197364. doi:10.3389/fimmu.2023.1197364
18. Tison A, Garaud S, Chiche L, Cornec D, Kostine M. Immune-checkpoint inhibitor use in patients with cancer and pre-existing autoimmune diseases. Nat Rev Rheumatol. 2022;18(11):641-656. doi:10.1038/s41584-022-00841-0
19. Hammad H, Debeuf N, Aegerter H, Brown AS, Lambrecht BN. Emerging Paradigms in Type 2 Immunity. Annu Rev Immunol. 2022;40:443-467. doi:10.1146/annurev-immunol-101320-030339
20. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 2009;10(8):864-871. doi:10.1038/ni.1770
21. Song W, Craft J. T Follicular Helper Cell Heterogeneity. Annu Rev Immunol. 2024;42(1):127-152. doi:10.1146/annurev-immunol-090222-102834
22. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015;74(1):5-17. doi:10.1016/j.cyto.2014.09.011
23. Park E, Ciofani M. Th17 cell pathogenicity in autoimmune disease. Exp Mol Med. 2025;57(9):1913-1927. doi:10.1038/s12276-025-01535-9
24. Zambrano-Zaragoza JF, Romo-Martínez EJ, Durán-Avelar Mde J, García-Magallanes N, Vibanco-Pérez N. Th17 cells in autoimmune and infectious diseases. Int J Inflam. 2014;2014:651503. doi:10.1155/2014/651503
25. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531-564. doi:10.1146/annurev.immunol.25.022106.141623
26. Grover P, Goel PN, Greene MI. Regulatory T Cells: Regulation of Identity and Function. Front Immunol. 2021;12:750542. doi:10.3389/fimmu.2021.750542
27. Takeuchi A, Saito T. CD4 CTL, a Cytotoxic Subset of CD4+ T Cells, Their Differentiation and Function. Front Immunol. 2017;8:194. doi:10.3389/fimmu.2017.00194
28. Koh CH, Lee S, Kwak M, Kim BS, Chung Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp Mol Med. 2023;55(11):2287-2299. doi:10.1038/s12276-023-01105-x
29. Wu L, Van Kaer L. Natural killer T cells in health and disease. Front Biosci (Schol Ed). 2011;3(1):236-251. doi:10.2741/s148
Mast Cells
Mast cells are present in mucosal tissues and connective tissues throughout the body and are functionally similar to basophils circulating in the blood (1).
Mucosal mast cell granules contain tryptase and chondroitin sulfate; connective tissue mast cell granules contain tryptase, chymase, and heparin (2). By releasing these mediators, mast cells play a key role in generating protective acute inflammatory responses. Mast cells also facilitate tissue repair and wound healing and promote physiologic homeostatic functions (3).
Basophils and mast cells are the source of type I hypersensitivity reactions associated with atopic allergy (4). They have high-affinity receptors for IgE called Fc-epsilon RI (FcεRI). Mast cell or basophil degranulation can be triggered by cross-linking of IgE receptors or by the anaphylatoxin complement fragments C3a and C5a.
Mast cells references
1. Pahima HT, Dwyer DF. Update on mast cell biology. J Allergy Clin Immunol. 2025;155(4):1115-1123. doi:10.1016/j.jaci.2024.12.1092
2. St John AL, Rathore APS, Ginhoux F. New perspectives on the origins and heterogeneity of mast cells. Nat Rev Immunol. 2023;23(1):55-68. doi:10.1038/s41577-022-00731-2
3. Boyce JA. Mast cells: beyond IgE. J Allergy Clin Immunol. 2003;111(1):24-33. doi:10.1067/mai.2003.60
4. Theoharides TC, Valent P, Akin C. Mast Cells, Mastocytosis, and Related Disorders. N Engl J Med. 2015;373(19):1885-1886. doi:10.1056/NEJMc1510021
Natural Killer (NK) Cells
Typical natural killer (NK) cells belong to a category of cells collectively referred to as innate lymphoid cells (which also includes ILC1, ILC2, and ILC3). They are best characterized by the expression of CD2+, CD8+, CD16+ (a receptor for IgG-Fc), and CD56+ surface markers and their lack of expression of CD3 and CD4-.
NK cells constitute 5 to 15% of peripheral blood mononuclear cells and have a round nucleus and granular cytoplasm (1). They induce apoptosis in infected or abnormal cells by a number of pathways. Like other innate lymphoid cells, they lack antigen-specific receptors; however, some NK cells have a form of immunologic memory.
Typical NK cells are thought to be important for tumor surveillance and immunity against viral infections (2). NK cells express both activating and inhibitory receptors. The activating receptors on NK cells can recognize numerous ligands on target cells (eg, MHC class I–related chain A [MICA] and chain B [MICB]). The inhibitory receptors on NK cells recognize MHC class I molecules. NK cells can kill their target only when there is no strong signal from inhibitory receptors. The presence of MHC class I molecules (normally expressed on nucleated cells) therefore prevents destruction of cells; their absence indicates that the cell is infected with certain viruses that downregulate MHC expression or has undergone malignant transformation with subsequent loss of MHC expression.
NK cells can also secrete several cytokines (eg, IFN-gamma, IL-1, TNF-alpha); they are a major source of IFN-gamma. By secreting IFN-gamma, NK cells can influence the acquired immune system by promoting the differentiation of type 1 helper T (Th1) cells and inhibiting that of type 2 helper (Th2) cells.
Patients with NK-cell deficiencies (eg, some types of severe combined immunodeficiency) are especially susceptible to herpesvirus infections and human papillomavirus infections, and NK cells may play either protective or pathogenic roles in the development of autoimmune disease (3).
Natural killer cells references
1. Mujal AM, Delconte RB, Sun JC. Natural Killer Cells: From Innate to Adaptive Features. Annu Rev Immunol. 2021;39:417-447. doi:10.1146/annurev-immunol-101819-074948
2. Wolf NK, Kissiov DU, Raulet DH. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol. 2023; 23:90-105. doi:10.1038/s41577-022-00732-1
3. Yang Y, Day J, Souza-Fonseca Guimaraes F, Wicks IP, Louis C. Natural killer cells in inflammatory autoimmune diseases. Clin Transl Immunology. 2021;10(2):e1250. doi:10.1002/cti2.1250
Polymorphonuclear Leukocytes
Polymorphonuclear leukocytes, also called granulocytes because their cytoplasm contains granules, include:
Neutrophils
Eosinophils
Basophils
Polymorphonuclear leukocytes occur in the circulation and have multilobed nuclei.
Monocyte
Eosinophil
Neutrophil
Basophil
Red blood cell (erythrocyte)
Lymphocyte
KATERYNA KON/SCIENCE PHOTO LIBRARY
Neutrophils
Neutrophils constitute 40 to 70% of total circulating white blood cells (1). they are the body's first line of defense against infection. Mature neutrophils are typically short-lived (ranging from a few hours to up to 5 days).
During acute inflammatory responses (eg, to infection), neutrophils, drawn by chemotactic factors, use adhesion molecules on blood vessel endothelium to leave the circulation and enter tissues. Their primary function is to phagocytose and digest pathogens. Microorganisms are killed when phagocytosis generates lytic enzymes and reactive oxygen compounds (eg, superoxide, hypochlorous acid) and triggers release of granule contents (eg, defensins, proteases, bactericidal permeability-increasing protein, lactoferrin, lysozymes). DNA and histones are also released, and they, with granule contents such as elastase, generate fibrous structures called neutrophil extracellular traps (NETs) in the surrounding tissues; these structures facilitate killing by trapping bacteria and focusing enzyme activity (2). Neutrophil extracellular traps also play a role in both facilitating and resolving inflammation (3, 4).
Patients with immunodeficiencies that affect the ability of phagocytes to kill pathogens (eg, chronic granulomatous disease) are especially susceptible to chronic bacterial and fungal infections.
Eosinophils
Eosinophils constitute up to 5% of circulating white blood cells (5).
They target organisms too large to be engulfed. Eosinophils kill by secreting toxic substances (eg, reactive oxygen compounds similar to those produced in neutrophils), major basic protein (which is toxic to parasites), eosinophil cationic protein (has cytotoxic activity against parasites, bacteria, and viruses but may also mediate inflammatory responses in allergic diseases), and several enzymes.
Circulating eosinophils, which increase in response to invasive parasitic infections, some cancers, and as part of an allergic response, are also a major source of inflammatory mediators (eg, prostaglandins, leukotrienes, platelet-activating factor, many cytokines). Because of their role as inflammatory mediators, eosinophils are implicated in a number of human diseases, including eosinophilic asthma, eosinophilic granulomatosis with polyangiitis, eosinophilic esophagitis, and hypereosinophilic syndrome.
Basophils
Basophils constitute < 5% of circulating white blood cells (6). Basophils share several characteristics with mast cells, although the 2 cell types have distinct lineages. Both have high-affinity receptors for IgE called Fc-epsilon RI (FcεRI). When these cells encounter certain antigens, the bivalent IgE molecules bound to the receptors become cross-linked, triggering cell degranulation with release of preformed inflammatory mediators (eg, histamine, platelet-activating factor) and the generation of newly synthesized mediators (eg, leukotrienes, prostaglandins, thromboxanes).
Polymorphonuclear leukocytes references
1. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The Neutrophil. Immunity. 2021;54(7):1377-1391. doi:10.1016/j.immuni.2021.06.006
2. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532-1535. doi:10.1126/science.1092385
3. Huang SU, O'Sullivan KM. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. Int J Mol Sci. 2022;23(7):3793. doi:10.3390/ijms23073793
4. Schoen J, Euler M, Schauer C, et al. Neutrophils' Extracellular Trap Mechanisms: From Physiology to Pathology. Int J Mol Sci. 2022;23(21):12855. Published 2022 Oct 25. doi:10.3390/ijms232112855
5. Arnold IC, Munitz A. Spatial adaptation of eosinophils and their emerging roles in homeostasis, infection and disease. Nat Rev Immunol. 2024;24(12):858-877. doi:10.1038/s41577-024-01048-y
6. Zhang N, Zhang Z-M, Wang X-F. The roles of basophils in mediating the immune responses. European Journal of Inflammation. 2021;19. doi:10.1177/20587392211047644





