



Spina bifida is defective closure of the vertebral column. Although the cause is often unknown, low folate levels during pregnancy increase risk. Some children are asymptomatic, and others have severe neurologic dysfunction below the lesion. Open spina bifida can be diagnosed prenatally by ultrasound or suggested by elevated alpha-fetoprotein levels in maternal serum and amniotic fluid. Postnatally, a lesion is typically visible on the back. Treatment is usually surgical.
Spina bifida is one of the most serious neural tube defects compatible with prolonged life. This defect is one of the more common congenital anomalies overall, with an incidence each year in the United States of about 1/2875 births (1). It is most common in the lower thoracic, lumbar, or sacral region and usually extends for 3 to 6 vertebral segments.
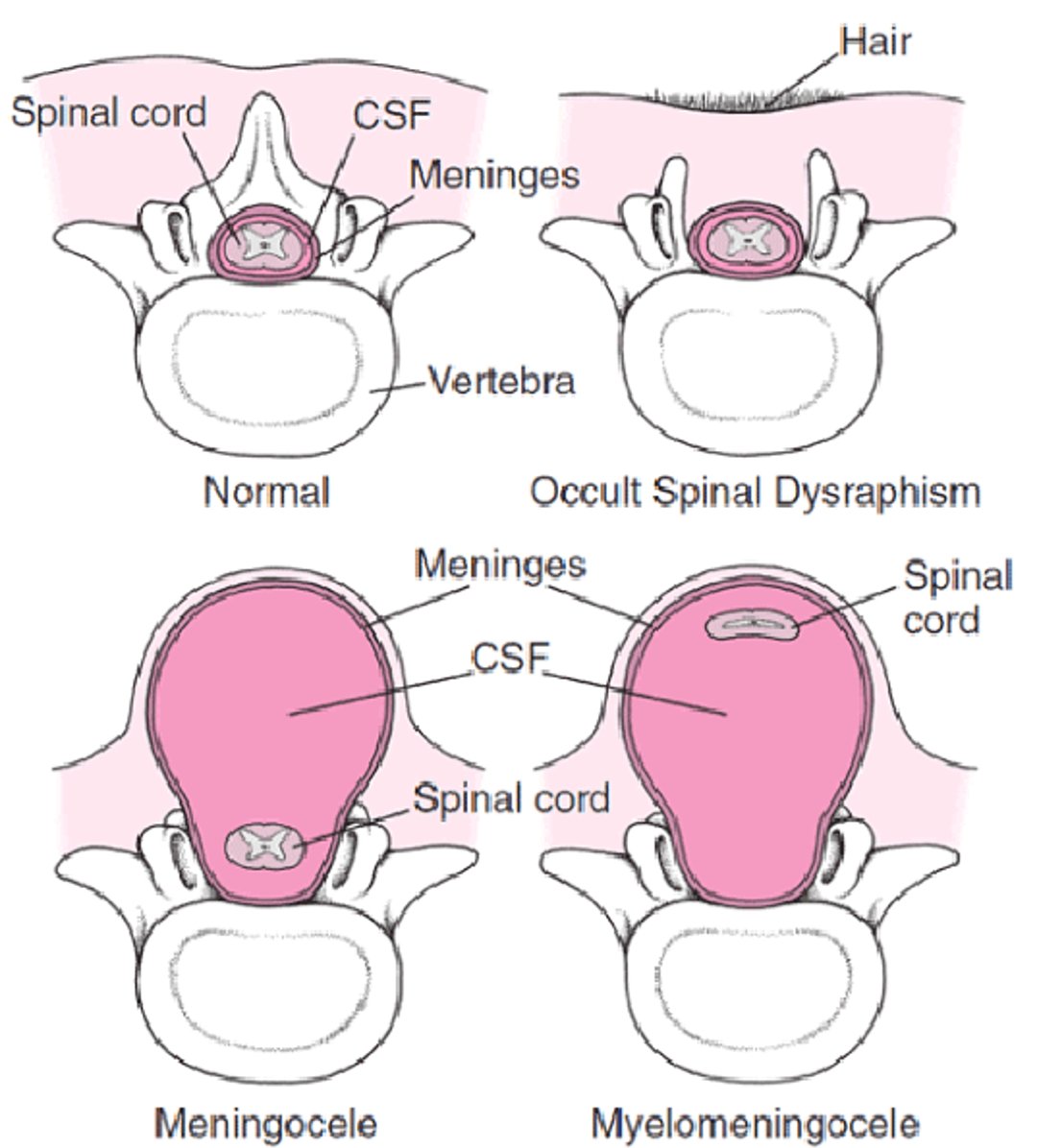
Severity ranges from occult (spina bifida occulta), in which there are no apparent anomalies or perhaps only skin discoloration or a tuft of hair over the area, to a protruding sac (spina bifida cystica), to a completely open spine (rachischisis) with severe neurologic disability and death.
In occult spinal dysraphism (OSD), anomalies of the skin overlying the lower back (typically in the lumbosacral area) occur. These anomalies include sinus tracts that have no visible bottom or are not in the midline; hyperpigmented areas; asymmetry of the gluteal cleft with the upper margin deviated to one side; sacral dimple or pit; and tufts of hair. These children often have anomalies in the underlying portion of the spinal cord, such as lipomas and tethering (in which the cord has an abnormal attachment).
In spina bifida cystica, the protruding sac can contain meninges (meningocele), spinal cord (myelocele), or both (myelomeningocele) (see figure ). In a myelomeningocele, the sac usually consists of meninges with a central neural plaque. If not well covered with skin, the sac can easily rupture, increasing the risk of meningitis.
Forms of Spina Bifida
In occult spinal dysraphism, ≥ 1 vertebrae do not form normally, and the spinal cord and meninges may also be affected. In spina bifida cystica, the protruding sac can contain meninges (meningocele), spinal cord (myelocele), or both (myelomeningocele). In a myelomeningocele, the meninges are usually exposed or, rarely, are covered by a thin layer of skin (as shown in the figure). |

Hydrocephalus is common because many children with spina bifida have a Chiari II type malformation.
Syringomyelia (a dilation of the normally small fluid-filled central canal of the spinal cord) and other congenital anomalies and soft-tissue masses around the spinal cord may be present.
General reference
1. Centers for Disease Control and Prevention: Data & Statistics on Spina Bifida. May 15, 2024. Accessed June 2025.
Etiology of Spina Bifida
Causes of spina bifida seem multifactorial. Folate deficiency is a significant factor, and there seems to be a genetic component.
Other risk factors include maternal use of certain medications (eg, valproate) and maternal diabetes.
Symptoms and Signs of Spina Bifida
Many children with minor defects are asymptomatic.
Neurologic
When the spinal cord or lumbosacral nerve roots are involved, as is usual, varying degrees of paralysis and sensory deficits are present below the lesion. Rectal tone is usually decreased.
Hydrocephalus may cause minimal symptoms or signs of increased intracranial pressure.
Brain stem involvement can be associated with hydrocephalus or syringomyelia. It may cause manifestations such as stridor, swallowing difficulties, and intermittent apnea.
Orthopedic
Lack of muscle innervation leads to atrophy of the legs. Because paralysis occurs in the fetus, orthopedic problems may be present at birth (eg, clubfoot, arthrogryposis of the legs, dislocated hip).
Kyphosis is sometimes present and can hinder surgical closure and prevent the child from lying supine. Scoliosis may develop later and is more common among children with higher lesions (ie, above L3).
Urologic
Paralysis also impairs bladder function, occasionally leading to a neurogenic bladder and, consequently, urinary reflux, which can cause hydronephrosis, frequent urinary tract infections, and, ultimately, kidney damage.
Diagnosis of Spina Bifida
Ultrasound or MRI
Spinal cord imaging, with ultrasound or MRI, is essential in children with occult spinal dysraphism; even children with minimal cutaneous findings may have underlying spinal abnormalities (those with overt defects do not require spinal cord imaging because the anatomy is known).
Plain radiographs of the spine, hips, and, if they are malformed, lower extremities, are done.
Cranial imaging using ultrasound, CT, or MRI is done to look for hydrocephalus and syringomyelia.
Once the diagnosis of spina bifida is made, urinary tract evaluation is essential and includes urinalysis, urine culture, blood urea nitrogen and creatinine determination, and ultrasound. Measurement of bladder capacity and pressure at which urine exits into the urethra can determine prognosis and intervention.
Need for further testing, such as urodynamics and voiding cystourethrogram, depends on previous findings and associated anomalies.
Screening
Prenatal screening can be done by doing fetal ultrasound and by measuring maternal serum levels of alpha-fetoprotein (see Maternal serum screening for neural tube defects), ideally between 16 weeks and 18 weeks gestation; levels of alpha-fetoprotein can also be measured in amniotic fluid samples if previous serum levels suggest an increased risk of a neural tube defect. Elevated levels suggest increased risk of spina bifida cystica (occult spinal dysraphism rarely causes elevated levels).
Treatment of Spina Bifida
Surgical repair of the spinal lesion (including fetal surgery)
Sometimes a ventricular shunt
Various measures for orthopedic and urologic complications
Without early surgical treatment, neurologic damage can progress in occult spinal dysraphism. Treatment for all forms of spina bifida requires a united effort by specialists from several disciplines; neurosurgical, urologic, orthopedic, pediatric, psychiatric, and social work evaluations are important. It is important to assess the type, vertebral segment, and extent of the lesion; the infant’s health status; and associated anomalies. Discussion with the family should ascertain the family’s strengths, desires, and resources and community resources, including availability of ongoing care.
A myelomeningocele identified prenatally may be repaired surgically before birth. Open and fetoscopic myelomeningocele procedures have become more widely used. Fetal surgery may be considered if the mother is relatively healthy and has normal prenatal testing results and if there is only one fetus with a myelomeningocele between T1 and S1. This surgery should be performed before viable preterm delivery age or gestational age < 25 6/7 weeks. Improved fetal outcomes of this surgery include delaying or avoiding the need to divert or shunt cerebrospinal fluid and decreased risk of infection; maternal outcomes improve as well (1).
A myelomeningocele identified perinatally is covered immediately after birth with a sterile dressing. If the myelomeningocele is leaking cerebrospinal fluid, antibiotics are started to prevent meningitis. Neurosurgical repair of a myelomeningocele or an open spine typically is done within the first 72 hours after birth to reduce the risk of meningeal or ventricular infection. If the lesion is large or is in a difficult location, plastic surgeons may be consulted to ensure adequate closure.
Hydrocephalus may require a shunt procedure in the neonatal period (see Treatment of Congenital Hydrocephalus); sometimes a ventricular shunt is inserted when the back is repaired.
Kidney function must be monitored closely, and urinary tract infection should be treated promptly. Obstructive uropathy at either the bladder outlet or ureteral level must be treated vigorously to prevent infection. When children are between 2 years and 3 years of age, or at any time if they have elevated pressure in the bladder with vesicoureteral reflux, clean intermittent catheterization is done to empty the bladder on a regular basis. Catheterization increases continence and maintains bladder and kidney health. (See also How To Do Pediatric Urologic Procedures.)
At around the same time, children are placed on the commode or toilet after meals to encourage fecal continence. Well-balanced diets are encouraged; stool softeners, laxatives, or a combination may be helpful to ensure regular bowel movements and to increase continence (see Treatment of Stool Incontinence). In older children, an antegrade colonic enema procedure, which is infusion of liquids through a gastrostomy tube, can improve continence.
Orthopedic care should begin early. If a clubfoot is present, a cast is applied; surgery is sometimes necessary after casting. Hip joints are checked for dislocation. Affected children should be monitored for development of scoliosis, pathologic fractures, pressure injuries, and muscle weakness and spasm.
Treatment reference
1. Kundishora AJ, Bond K, Rosenfeld M, et al. Detailed analysis of hydrocephalus patterns and associated variables in patients after open fetal repair and postnatal myelomeningocele/myeloschisis closure. Childs Nerv Syst. 2025;41(1):160. Published 2025 Apr 16. doi:10.1007/s00381-025-06819-z
Prognosis for Spina Bifida
Prognosis varies by the level of cord involvement and the number and severity of associated anomalies. Prognosis is worse for children with higher cord level (eg, thoracic) lesions or who have kyphosis, hydrocephalus, early hydronephrosis, and associated congenital anomalies. With proper care, however, most children do well.
Loss of renal function and ventricular shunt complications are the usual causes of death in older children.
Prevention of Spina Bifida
Folate supplementation (400 to 800 mcg orally once/day) in women beginning 3 months before conception and continuing through the first trimester reduces the risk of neural tube defects (see prevention of congenital neurologic anomalies).
Women who have had a fetus or infant with a neural tube defect are at high recurrence risk and should take folate 4 mg (4000 mcg) orally once/day beginning 3 months before conception and continuing through the first trimester.
Key Points
Spina bifida involves defective closure of the vertebral column, sometimes with a protruding sac containing meninges (meningocele), spinal cord (myelocele), or both (myelomeningocele).
Chiari II type malformation, often causing hydrocephalus, is common.
Folate deficiency is a significant risk factor, but other factors include maternal use of certain medications (eg, valproate), maternal diabetes, and possibly a genetic component.
Children with minor defects can be asymptomatic, but others typically have varying degrees of paralysis and sensory deficits below the lesion.
Lack of muscle innervation leads to atrophy of the legs and orthopedic deformities.
Screen prenatally using fetal ultrasound and measuring maternal serum levels of alpha-fetoprotein.
Repair the spinal lesion, place a shunt for symptomatic hydrocephalus, and treat orthopedic and urologic abnormalities as needed; consider prenatal surgical repair in selected patients.
Reduce risk by giving folate supplementation beginning before conception.
Drug Information for the Topic





