



Duchenne muscular dystrophy and Becker muscular dystrophy are X-linked recessive disorders characterized by progressive proximal muscle weakness caused by muscle fiber degeneration. Becker dystrophy has later onset and causes milder symptoms. Diagnosis is suggested clinically and is confirmed by genetic testing or analysis of the protein product (dystrophin) of the mutated gene. Treatment is focused on maintaining function through physical therapy and the use of braces and orthotics. Patients who have Duchenne dystrophy should be offered prednisone or deflazacort and sometimes genetic therapies.
Muscular dystrophies are inherited, progressive muscle disorders resulting from defects in one or more genes needed for normal muscle structure and function; dystrophic changes (eg, muscle fiber necrosis and regeneration) are seen on biopsy specimens.
Duchenne dystrophy and Becker dystrophy are the most prevalent muscular dystrophies. They are caused by mutations of the dystrophin gene, the largest known human gene, at the Xp21.2 locus. Up to 70% of Duchenne dystrophy is caused by a single- or multiexon deletion, approximately 10% by a duplication and 20% by a point mutation. In Becker dystrophy, approximately 70% of patients have a deletion, 20% have a duplication, and up to 10% are point mutations (1).
In Duchenne dystrophy, these mutations result in the severe absence (< 5%) of dystrophin, a protein in the muscle cell membrane. In Becker dystrophy, the mutations result in production of abnormal dystrophin or insufficient dystrophin.
Duchenne dystrophy and Becker dystrophy together affect about 1/5000 to 1/6000 live male births (1); the majority have Duchenne. Female carriers may have asymptomatic elevated creatine kinase levels and possibly calf hypertrophy.

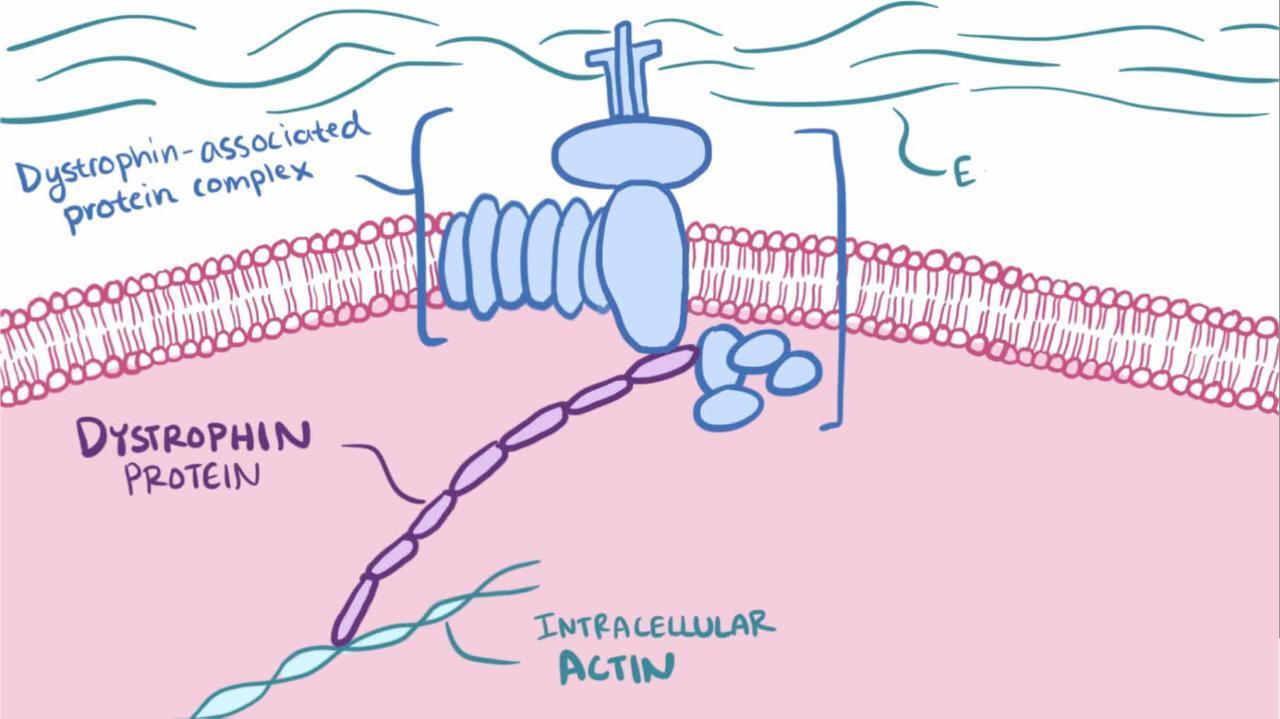
General reference
1. Duan D, Goemans N, Takeda S, et al: Duchenne muscular dystrophy. Nat Rev Dis Primers 7(1):13, 2021. doi: 10.1038/s41572-021-00248-3
Symptoms and Signs
Duchenne dystrophy
This disorder affects approximately 20/100,000 live male births and manifests typically between 2 and 3 years of age (1). Weakness affects proximal muscles, typically in the lower limbs initially. Children frequently toe walk and have a waddling gait and lordosis. They have difficulty running, jumping, climbing stairs, and rising from the floor. Children fall frequently, often causing arm or leg fractures (in approximately 20% of patients) (2). Progression of weakness is steady, and limb flexion contractures and scoliosis develop in nearly all children. Firm pseudohypertrophy (fatty and fibrous replacement of certain enlarged muscle groups, notably the calves) develops. Most children need to use a wheelchair by age 12, and, if they are unsupported by mechanical ventilation, most die of respiratory complications by age 20. Children who are supported with ventilation may live an additional 10 to 20 years.
Consequences of cardiac muscle involvement include dilated cardiomyopathy, conduction abnormalities, and arrhythmias. Such complications occur in about one third of patients by age 14 and in all patients over age 18; however, because these patients are not able to exercise, cardiac involvement is usually asymptomatic until late in the disease. About one third have mild, nonprogressive intellectual impairment that affects verbal ability more than performance.
Becker dystrophy
Compared to Duchenne dystrophy, Becker dystrophy affects < 8/100,000 live male births, typically becomes symptomatic much later, and is milder (3). Ambulation is usually preserved until at least age 15, and many children remain ambulatory into adulthood. Most affected children survive into their 30s and 40s.
Symptoms and signs references
1. Kariyawasam D, D'Silva A, Mowat D, et al: Incidence of Duchenne muscular dystrophy in the modern era; an Australian study. Eur J Hum Genet 30(12):1398-1404, 2022. doi: 10.1038/s41431-022-01138-2
2. McDonald DG, Kinali M, Gallagher AC, et al: Fracture prevalence in Duchenne muscular dystrophy. Dev Med Child Neurol 44(10):695-698, 2002. doi: 10.1017/s0012162201002778
3. Duan D, Goemans N, Takeda S, et al: Duchenne muscular dystrophy. Nat Rev Dis Primers 7(1):13, 2021. doi: 10.1038/s41572-021-00248-3
Diagnosis
DNA mutation analysis
Sometimes muscle biopsy with immunostaining analysis of dystrophin
Diagnosis is suspected by characteristic clinical findings, age at onset, and family history suggestive of X-linked recessive inheritance. Myopathic changes are noted on electromyography (rapidly recruited, short duration, low-amplitude motor unit potentials) and, when done, muscle biopsy shows necrosis and marked variation in muscle fiber size not segregated by motor unit. Creatinine kinase levels are elevated up to 100 times normal.
Mutation analysis of DNA from peripheral blood leukocytes using multiplex ligation-dependent probe amplification (MLPA) is the primary confirmatory test; it can identify abnormalities in the dystrophin gene. If an abnormality is not detected by MLPA but Duchenne or Becker dystrophy is still suspected, full sequencing of the dystrophin gene can be done to detect small genetic changes, such as point mutations.
If genetic testing does not confirm the diagnosis, then analysis of dystrophin with immunostaining of muscle biopsy samples should be done. Dystrophin is undetectable in patients with Duchenne dystrophy. In patients with Becker dystrophy, dystrophin is typically abnormal (lower molecular weight) or present in low concentration.
Patients with Duchenne dystrophy should have a baseline assessment of cardiac function with ECG and echocardiography at the time of diagnosis or by age 6 years.
Carrier detection and prenatal diagnosis are possible by using conventional studies (eg, pedigree analysis, creatinine kinase determinations, fetal sex determination) combined with recombinant DNA analysis and dystrophin immunostaining of muscle tissue.
Treatment
Supportive measures
Sometimes corrective surgery
Sometimes, for cardiomyopathy, an angiotensin-converting enzyme inhibitor and/or beta-blocker
For Duchenne dystrophy, prednisone or deflazacort and sometimes genetic therapy
Treatment involves a multidisciplinary approach that includes both nonpharmacologic and pharmacologic measures, which include genetic therapies. Gentle (ie, submaximal) active exercise is encouraged for as long as possible to avoid disuse atrophy or complications of inactivity. Passive exercises may extend the period of ambulation. Orthopedic interventions should be aimed at maintaining function and preventing contractures. Ankle-foot orthoses worn during sleep may help prevent flexion contractures. Leg braces may temporarily help preserve ambulation or standing. Corrective surgery is sometimes needed, particularly for scoliosis. Obesity should be avoided; caloric requirements are likely to be less than normal because of decreased physical activity.
Respiratory insufficiency may be treated with noninvasive ventilatory support (eg, nasal mask) and sometimes with mechanical ventilation. Elective tracheotomy is gaining acceptance, allowing children with Duchenne dystrophy to live into their 30s and beyond.
For children with dilated cardiomyopathy, an angiotensin-converting enzyme inhibitor and/or a beta-blocker may help prevent or slow progression (1).
Investigational therapies for Duchenne dystrophy and Becker dystrophy include gene therapy, creatine, myostatin inactivation, skeletal muscle progenitors, and the antioxidant idebenone (2).
Genetic counseling is indicated.
Corticosteroids for Duchenne dystrophy
In Duchenne dystrophy, daily corticosteroids (prednisone or deflazacort) are the mainstay of therapy for patients > age 4 years who are no longer gaining or have declining motor skills (3). Corticosteroids start working as early as 10 days after initiation of therapy; efficacy peaks at 3 months and persists for 6 months. Long-term use improves strength, delays the age at which ambulation is lost by 1.4 to 2.5 years, improves timed function testing (a measurement of how fast a child completes a functional task, such as walking or getting up from the floor), improves pulmonary function, reduces orthopedic complications (eg, the need for scoliosis surgery), stabilizes cardiac function (eg, delays onset of cardiomyopathy until 18 years of age), and increases survival by 5 to 15 years (3). Alternate-day prednisone is not effective. Weight gain and cushingoid facies are common adverse effects after 6 to 18 months. Risk of vertebral compression and long bone fractures also is increased.
Deflazacort may be associated with a greater risk of cataracts than prednisone.
Use of prednisone or deflazacort in Becker dystrophy has not been adequately studied.
Genetic therapy options for Duchenne dystrophy
Genetic therapies that increase dystrophin levels are available in some countries, but they are costly and their benefit is uncertain (4). The use of these therapies requires careful consideration and shared-decision making.
Exon-skipping medications (IV eteplirsen, golodirsen, viltolarsen, and casimersen) use antisense oligonucleotides that work like molecular patches to the abnormal dystrophin gene in which 1 or more exons are missing (the missing exons prevent the full protein from being assembled thus causing severe symptoms). These medications mask an exon so that it will be skipped and ignored during protein production, allowing for the production of a dystrophin protein that, although not normal, is functional and theoretically lessens symptoms so that patients function more like boys with the less severe Becker muscular dystrophy.
Eteplirsen skips exon 51. Limited data suggest that eteplirsen leads to increased dystrophin in muscle and increased walking performance on timed tests in the 13% of patients with Duchenne dystrophy who have a dystrophin gene mutation that is amenable to exon 51 skipping. The medication's approval has been criticized because it was based on a small trial that relied on a surrogate outcome (dystrophin in muscle biopsy) (5), and clinical benefit remains unproved.
Golodirsen and viltolarsen skip exon 53. They can be used in the 8% of patients with Duchenne dystrophy who have a mutation in the dystrophin gene amenable to exon 53 skipping. Clinical benefit remains unproved.
Casimersen skips exon 45. It can be used in the 8% of patients with Duchenne dystrophy with a confirmed mutation amenable to exon 45 skipping. It increases dystrophin production, but clinical benefit has not been shown.
Stop codon read-through medications (eg, oral ataluren [PTC124]) bypass premature stop codons, allowing for the production of a functional protein. Stop codons are nonsense mutations that stop the production of a functioning protein too early, resulting in a truncated, nonfunctional protein.
Ataluren promotes ribosomal read-through of premature but not normal termination codons and aims to produce a functional dystrophin protein. It is an option for patients with Duchenne dystrophy who are 2 years of age and older, who are ambulatory, and whose disease is caused by nonsense mutations. Ataluren is available in the European Union and United Kingdom. Clinical benefit is unproved (6).
Gene transfer via viral vectors (eg, delandistrogene moxeparvovec) use viral vectors to deliver corrective genetic material to the affected muscles.
Despite the lack of proven clinical benefit, delandistrogene moxeparvovec is the only agent approved for gene transfer therapy via viral vector, whereby microdystrophin transgenes can be delivered to skeletal and cardiac muscle using an adeno-associated virus capsid (AAVrh74). No functional improvement was seen in the trial (7), but in a subgroup analysis, some improvement in functional status was seen in the 4- to 5-year-old age group (8). Adverse effects include nausea, vomiting, fever, hepatic dysfunction, and thrombocytopenia as well as life-threatening immune inflammatory reactions. Further study is needed to determine this agent's place in the therapeutic armamentarium for Duchenne dystrophy.
Treatment references
1. Birnkrant DJ, Bushby K, Bann CM, et al: Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 17(4):347-361, 2018. doi: 10.1016/S1474-4422(18)30025-5
2. Ren S, Yao C, Liu Y, et al: Antioxidants for Treatment of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Eur Neurol 85(5):377-388, 2022. doi: 10.1159/000525045
3. Gloss D, Moxley RT 3rd, Ashwal S, Oskoui M: Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 86:465–472, 2016. doi: 10.1212/WNL.0000000000002337
4. Bendicksen L, Zuckerman DM, Avorn J, et al: The Regulatory Repercussions of Approving Muscular Dystrophy Medications on the Basis of Limited Evidence. Ann Intern Med 176(9):1251-1256, 2023. doi: 10.7326/M23-1073
5. Mendell JR, Goemans N, Lowes LP, et al: Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79(2):257-271, 2016. doi: 10.1002/ana.24555
6. McDonald CM, Campbell C, Torricelli RE, et al: Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:(10101):1489–1498, 2017. doi: 10.1016/S0140-6736(17)31611-2
7. Mendell JR, Shieh PB, McDonald CM, et al: Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front Cell Dev Biol 11:1167762, 2023. doi: 10.3389/fcell.2023.1167762
8. Elevidys. Prescribing information. Sarepta Therapeutics, Inc; 2023. Accessed January 9, 2024.
Key Points
Duchenne dystrophy and Becker dystrophy are X-linked recessive disorders that cause a decrease in dystrophin, a protein in muscle cell membranes.
Patients have significant, progressive weakness that causes severe disability, including difficulty walking, frequent falls, dilated cardiomyopathy, and early death due to respiratory insufficiency.
Active and passive exercise is helpful, along with leg braces and ankle-foot orthoses.
In Duchenne dystrophy, daily prednisone or deflazacort improves muscle strength and mass, improves pulmonary function, and helps delay onset of cardiomyopathy, although adverse effects are common.
For patients with Duchenne dystrophy with certain mutations, eteplirsen, golodirsen, viltolarsen, casimersen, and ataluren, despite limited evidence of clinical benefit, may be used as well.
An angiotensin-converting enzyme inhibitor and/or a beta-blocker may help prevent or slow progression of cardiomyopathy.
Ventilatory support (noninvasive and, later on, invasive) can help prolong life.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
Muscular Dystrophy Association: Information on research, treatment, technology, and support for patients living with Duchenne muscular dystrophy and Becker muscular dystrophy
National Organization for Rare Disorders: Comprehensive information regarding Duchenne muscular dystrophy and Becker muscular dystrophy, including standard and investigational therapies and links to related topics
Muscular Dystrophy News Today: A news and information web site about muscular dystrophy
Drug Information for the Topic





