



Neurofibromatosis refers to a group of several related genetic disorders that have overlapping clinical manifestations. It causes various types of benign or malignant tumors that involve central or peripheral nerves and often causes pigmented skin macules and sometimes other manifestations. Diagnosis is primarily clinical based on specific criteria. Treatment options for benign tumors include surgical intervention, targeted medical therapies (eg, MEK inhibitors and vascular endothelial growth factor inhibitors), and multidisciplinary supportive care. Malignant tumors (which are less common) can be treated with chemotherapy.
Neurofibromatosis is a group of genetic neurocutaneous syndromes that are characterized by the gradual development of benign and/or malignant tumors.
Epidemiology and Etiology of Neurofibromatosis
There are 3 main types of neurofibromatosis:
Neurofibromatosis type 1 (NF1)
NF2-related schwannomatosis (NF2)
Non-NF2 schwannomatosis (schwannomatosis)
Note that NF2-related schwannomatosis and non-NF2 schwannomatosis are terminologies recommended by the International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC) because they better reflect the updated criteria for these disorders that incorporate clinical features and genetic testing (1). (See also table .)
Neurofibromatosis type 1 (NF1)
NF1 (von Recklinghausen disease) is the most prevalent type. It accounts for approximately 96% of all neurofibromatoses (2). In one meta-analysis of global data, the pooled incidence of NF1 at birth was approximately 1 in 2600 patients (3).
NF1 is caused by germline pathogenic variants in NF1 (a tumor suppressor gene) located on band 17q11.2, which encodes the synthesis of neurofibromin; > 1000 such variants have been identified. Inheritance is autosomal dominant, and approximately 50% of cases can be caused by a de novo germ cell mutation (4).
NF1 causes neurologic, cutaneous, and sometimes soft-tissue or bone manifestations. Phenotypic variation is significant (5).
NF2-related schwannomatosis (NF2)
NF2 accounts for approximately 3% of all neurofibromatoses (2). In a study from the North West of England in the United Kingdom, birth incidence of NF2 was 1 in 27,956 (6).
NF2 is a tumor suppressor gene located on band 22q12.2 and encodes the synthesis of merlin (7); 200 variants have been identified. About half of patients with NF2 inherit a mutation from an affected parent in an autosomal dominant manner (8).
NF2 manifests primarily as congenital bilateral vestibular schwannomas (tumors on the vestibular branch of the eighth cranial nerve).
Non-NF2 schwannomatosis
Non-NF2 schwannomatosis (schwannomatosis) is the most rare type of neurofibromatosis. Schwannomatosis was previously considered a variant of NF2 because multiple schwannomas are present in both conditions; however, it has subsequently been recognized as a genetically and clinically distinct disorder.
In one study from the North West of England in the United Kingdom, birth incidence of schwannomatosis was 1 in 68,956 (6). In 15 to 25% of cases, schwannomatosis is familial and related to a germline mutation in the SMARCB1 or LZTR1 gene, and inheritance is autosomal dominant (9). Both of these are tumor suppressor genes and are located on chromosome 22q11.21-23 very close to the NF2 gene (1). In the remaining cases, the genetic basis is not well-understood, but in tissue from some patients, other mutations in the same gene are involved.
Schwannomas develop in spinal and peripheral nerves and can sometimes be quite painful. Unlike NF2, vestibular schwannomas typically do not develop in schwannomatosis.
Epidemiology and etiology references
1. Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24(9):1967-1977. doi:10.1016/j.gim.2022.05.007
2. Tamura R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int J Mol Sci. 2021;22(11):5850. Published 2021 May 29. doi:10.3390/ijms22115850
3. Lee TJ, Chopra M, Kim RH, Parkin PC, Barnett-Tapia C. Incidence and prevalence of neurofibromatosis type 1 and 2: a systematic review and meta-analysis. Orphanet J Rare Dis. 2023;18(1):292. Published 2023 Sep 14. doi:10.1186/s13023-023-02911-2
4. Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; October 2, 1998.
5. Peduto C, Zanobio M, Nigro V, Perrotta S, Piluso G, Santoro C. Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype-Phenotype Correlations. Cancers (Basel). 2023;15(4):1217. Published 2023 Feb 14. doi:10.3390/cancers15041217
6. Evans DG, Bowers NL, Tobi S, et al. Schwannomatosis: a genetic and epidemiological study. J Neurol Neurosurg Psychiatry. 2018;89(11):1215-1219. doi:10.1136/jnnp-2018-318538
7. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 607379: July 26, 2023. World Wide Web URL: www.omim.org
8. Evans DG, Huson SM, Donnai D, et al:. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29(12):841-846. doi:10.1136/jmg.29.12.841
9. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); [updated 2020 Jun 24]. Schwannomatosis; [updated 2017 Jan 1; reviewed 2018 Jun 01; cited 2025 Nov 11]; [about 5 p.]. Available from: Schwannomatosis: MedlinePlus Genetics.
Pathophysiology of Neurofibromatosis
Tumor development in neurofibromatoses is driven by loss of function in key tumor suppressor genes (NF1, NF2, SMARCB1, LZTR1), leading to dysregulation of cell signaling pathways, with additional genetic and microenvironmental factors modulating tumor initiation, growth, and malignant transformation. Neurofibromin is a tumor suppressor protein encoded by the NF1 gene, and merlin is a tumor suppressor protein encoded by the NF2 gene, which when mutated cause neurofibromatosis type 1 and type 2.
Important dysregulated pathways that cause tumors include (1):
NF1: Loss of neurofibromin functions leads to Ras/MAPK and PI3K/mTOR activation. driving uncontrolled cell proliferation.
NF2: Loss of merlin disrupts contact inhibition of PI3K/AKT, Raf/MEK/ERK, and mTOR signaling and allows uncontrolled Schwann cell proliferation, leading to bilateral vestibular schwannomas
Non-NF2 schwannomatosis: Inactivation of both schwannomatosis genes SMARCB1 and LZTR1 and of NF2 is responsible for the development of multiple nonvestibular schwannomas, which is in contrast to the bilateral vestibular schwannomas characteristic of NF2.
A proposed hypothesis for familial schwannomatosis suggests that tumor development is caused by 3 sequential genetic events:
A germline mutation in either the LZTR1 or SMARCB1 gene
Loss of heterozygosity on chromosome 22q that eliminates the wild-type alleles of LZTR1, SMARCB1, and NF2
A somatic mutation that inactivates the remaining NF2 allele (2)
This cascade of events is ultimately theorized to result in the biallelic inactivation of LZTR1, SMARCB1, and NF2.
Tumors may be peripheral or central.
Peripheral tumors are more common in NF1 and can develop anywhere along the course of the peripheral nerves. The tumors are histopathologically deemed neurofibromas, which develop from nerve sheaths and consist of mixtures of Schwann cells, fibroblasts, neural cells, and mast cells. Most appear during adolescence. Occasionally, they can transform to malignant peripheral nerve sheath tumors. There are multiple forms:
Cutaneous neurofibromas are soft and fleshy.
Subcutaneous neurofibromas are firm and nodular.
Nodular plexiform neurofibromas may involve spinal nerve roots, typically growing through an intervertebral foramen to cause intraspinal and extraspinal masses (dumbbell tumor). The intraspinal part may compress the spinal cord.
Diffuse plexiform neurofibromas (subcutaneous nodules or amorphous overgrowth of underlying bone or Schwann cells) can be disfiguring and may cause deficits distal to the neurofibroma. Diffuse plexiform neurofibromas can become malignant and they appear to be the most common precursors to malignant peripheral nerve sheath tumors in people with NF1.
Schwannomas are derived from Schwann cells, rarely undergo malignant transformation, and can occur in peripheral nerves anywhere in the body. Compared to the other peripheral tumors, schwannomas are rare in NF1.
Central tumors have several forms:
Optic gliomas: These tumors are low-grade pilocytic astrocytomas, which may be asymptomatic or may progress enough to compress the optic nerve and cause blindness. These tumors occur in younger children and can usually be identified by age 5 and rarely develop after age 10. They occur in NF1.
Vestibular schwannomas (acoustic neuromas): They tumors are the key distinguishing feature of NF2. They may cause dizziness, ataxia, deafness, and tinnitus due to compression of the eighth cranial nerve; they sometimes cause facial weakness due to compression of the adjacent seventh cranial nerve.
Meningiomas: These tumors develop in some people, particularly those with NF2.
Pathophysiology references
1. Tamura R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int J Mol Sci. 2021;22(11):5850. Published 2021 May 29. doi:10.3390/ijms22115850
2. Dhamija R, Plotkin S, Gomes A, Babovic-Vuksanovic D. LZTR1- and SMARCB1-Related Schwannomatosis. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; March 8, 2018.
Symptoms and Signs of Neurofibromatosis
Neurofibromatosis type 1 (NF1)
Most patients with NF1 are asymptomatic. Some present with neurologic symptoms or bone deformities. In many patients, characteristic skin lesions may be apparent at birth or develop during infancy.
Café-au-lait lesions are medium-brown (café-au-lait), freckle-like macules distributed most commonly over the trunk, pelvis, and flexor creases of elbows and knees. Children who have NF1 have ≥ 6 café-au-lait macules and often many more. These macules are > 5 mm in affected prepubertal children and > 15 mm in postpubertal patients (see table ). The number of such macules at birth directly correlates with the likelihood of having NF1 and is as high as 95% in patients with ≥ 5 macules (1). Café-au-lait lesions occur in conditions other than neurofibromatosis and should be included in the differential diagnosis (eg, Legius syndrome, McCune-Albright syndrome, Noonan syndrome, tuberous sclerosis complex, Fanconi anemia).
Cutaneous neurofibromas, which arise along small peripheral nerves, are common. During late childhood, these cutaneous tumors of various sizes and shapes appear, ranging in number from several to thousands. They may appear skin-colored or have a pink or tan discoloration and are usually asymptomatic.
Plexiform neurofibromas can develop and have a tendency to grow to large sizes, causing irregularly thickened, distorted structures, sometimes with grotesque deformities that can impinge on nerves and other structures. Plexiform neurofibromas can also involve cranial nerves, typically the fifth, ninth, and tenth.
Neurologic symptoms vary, depending on location and number of neurofibromas. Larger neurofibromas may press on their nerve of origin and cause distal paresthesia, pain, and sensory loss or weakness, depending on the function of the nerve that is involved. Neurofibromas that form along spinal nerve roots, especially where the nerve roots are contained by bone, can compress the nerve roots and cause radicular pain, weakness, or widespread sensory loss in that nerve distribution. Compression of cranial nerves by plexiform neurofibromas manifests as clinical symptoms consistent with impairment of the affected nerves (eg, pain, sensory deficits, motor deficits).


This photo shows a single neurofibroma, which is a benign tumor arising from the fibrous covering surrounding the nerves.
This photo shows a single neurofibroma, which is a benign tumor arising from the fibrous covering surrounding the nerve
DR P. MARAZZI/SCIENCE PHOTO LIBRARY

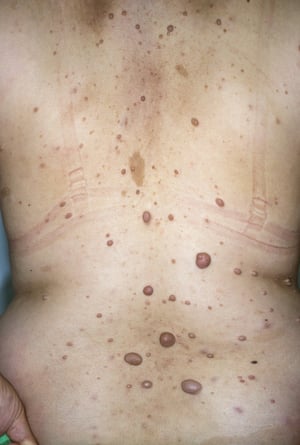
This photo shows multiple neurofibromas (raised pink or tan nodules) and café-au-lait spots (flat brownish spots) on the back of a patient with neurofibromatosis.
This photo shows multiple neurofibromas (raised pink or tan nodules) and café-au-lait spots (flat brownish spots) on th
DR HAROUT TANIELIAN/SCIENCE PHOTO LIBRARY

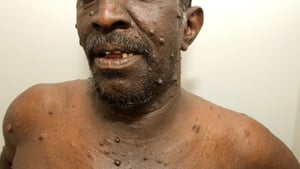
This photo shows multiple neurofibromas (tan nodules) on a patient with neurofibromatosis.
This photo shows multiple neurofibromas (tan nodules) on a patient with neurofibromatosis.
MEDICIMAGE / SCIENCE PHOTO LIBRARY

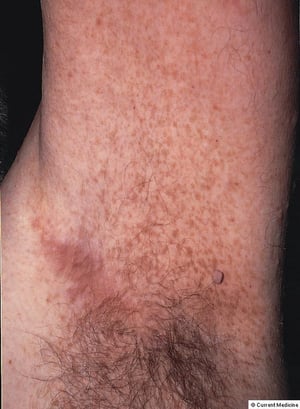
This photo shows axillary freckle-like macules in NF1.
This photo shows axillary freckle-like macules in NF1.
© Springer Science+Business Media
This photo shows a single neurofibroma, which is a benign tumor arising from the fibrous covering surrounding the nerves.
This photo shows a single neurofibroma, which is a benign tumor arising from the fibrous covering surrounding the nerve
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
This photo shows multiple neurofibromas (raised pink or tan nodules) and café-au-lait spots (flat brownish spots) on the back of a patient with neurofibromatosis.
This photo shows multiple neurofibromas (raised pink or tan nodules) and café-au-lait spots (flat brownish spots) on th
DR HAROUT TANIELIAN/SCIENCE PHOTO LIBRARY
This photo shows multiple neurofibromas (tan nodules) on a patient with neurofibromatosis.
This photo shows multiple neurofibromas (tan nodules) on a patient with neurofibromatosis.
MEDICIMAGE / SCIENCE PHOTO LIBRARY
This photo shows axillary freckle-like macules in NF1.
This photo shows axillary freckle-like macules in NF1.
© Springer Science+Business Media
Bone abnormalities include:
Dysplasia of the greater wing of the sphenoid bone (posterior orbital wall), with consequent pulsating exophthalmos
Subperiosteal bone cysts
Vertebral scalloping
Thinning of the long-bone cortex (eg, tibial bowing)
Pseudarthrosis


This coronal T2-weighted MR image shows sphenoid wing dysplasia and a large plexiform neurofibroma infiltrating the left scalp and masticator spaces.
Living Art Enterprises/SCIENCE PHOTO LIBRARY

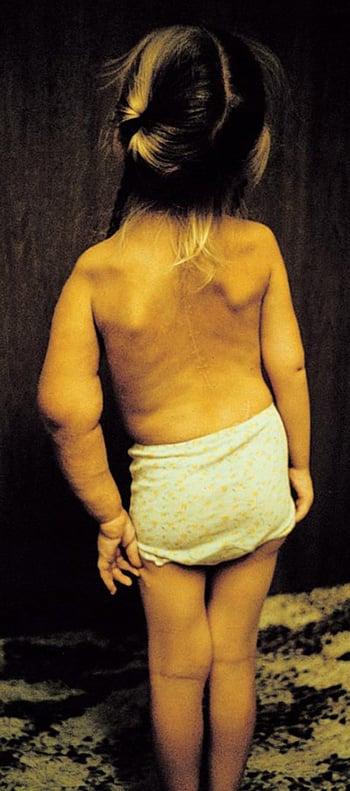
In this photo, the patient’s left arm has a plexiform neuroma extending from the deltoid proximal humerus to the hand. Her humerus is affected by multiple midshaft fractures associated with thinning of bone cortex and pseudoarthrosis. She also has scoliosis, short stature, and enlargement of the lumbar intraspinal canal caused by anterior meningocele.
An optic glioma and Lisch nodules (iris hamartomas) occur in some patients. Optic gliomas are typically asymptomatic and do not require treatment unless they progressively increase in size, which can result in blurred vision or loss of peripheral vision, double vision, and pain (2).
Patients with NF1 can also have changes in their arterial walls that may lead to Moyamoya syndrome (stenosis or occlusion of arteries in and around the circle of Willis with formation of small collateral arteries) or intracranial aneurysms.
Some children have cognitive deficits, learning disabilities, and macrocephaly.


Lisch nodules of the iris are hyperpigmented melanotic hamartomas (arrows).
Children and adolescents with NF1 may develop childhood chronic myelomonocytic leukemia, juvenile myelomonocytic leukemia, and rhabdomyosarcoma. Pheochromocytomas may occur at any age.
Malignant tumors are much less common but are more common than in the general population; they include supratentorial or brain stem gliomas and transformation of plexiform neurofibromas to malignant peripheral nerve sheath tumors. These tumors may develop at any age.
NF2-related schwannomatosis (NF2)
In NF2, bilateral vestibular schwannomas develop and become symptomatic during childhood or early adulthood. They cause bilateral hearing loss, problems of balance (eg, vertigo, unsteadiness), and sometimes headache or facial weakness. Bilateral eighth cranial (vestibulocochlear) nerve masses may be present.
Subcapsular or cortical cataract, retinal hamartoma, or epiretinal membrane may occur in childhood or young adulthood.
Family members may have gliomas (eg, ependymomas), meningiomas, or schwannomas.
Non-NF2 schwannomatosis (schwannomatosis)
In schwannomatosis, multiple schwannomas develop on cranial, spinal, and peripheral nerves. Notably, vestibular schwannomas do not develop, and patients do not develop hearing loss. The other types of tumors that sometimes occur in neurocutaneous disorders do not develop either.
The first symptom of schwannomatosis is usually pain, which may become chronic and severe. Other symptoms may develop, depending on the location of the schwannomas.
Symptoms and signs references
1. Santangelo A, Chelleri C, Tomasino M, et al. Café-Au-Lait Macules in Neurofibromatosis Type 1: Birthmark or Biomarker?. Cancers (Basel). 2025;17(9):1490. Published 2025 Apr 29. doi:10.3390/cancers17091490
2. Rasool N, Odel JG, Kazim M. Optic pathway glioma of childhood. Curr Opin Ophthalmol. 2017;28(3):289-295. doi:10.1097/ICU.0000000000000370
Diagnosis of Neurofibromatosis
Primarily history and physical examination
Brain MRI or head CT
Sometimes genetic testing
NF1 is most often suspected during a routine physical examination (sometimes prompted by cosmetic concerns), or during an evaluation based on a positive family history. The diagnosis of all 3 types is primarily clinical (see table ) by detailed physical examination that is focused on the cutaneous, skeletal, and neurologic systems. NF1 should be suspected and monitored for in children who have multiple café-au-lait spots even if they do not have other features or a family history of NF1 (eg, such as those with Legius syndrome) (1).
Imaging with brain MRI is performed in patients with neurologic symptoms or signs and, when detailed visual testing is not possible, in young children who meet the clinical criteria for NF1 and who may have an optic glioma. T2-weighted MRI may show thickening or tortuosity of the optic nerves and parenchymal hyperintense lesions that change over time and correlate with small cystic structures in NF1; MRI may help identify vestibular schwannomas or meningiomas in NF2 (2). If vestibular schwannoma is suspected, CT of the petrous ridge can be done; it typically shows widening of the auditory canal.
Although the diagnosis can usually be established by clinical criteria, genetic testing, specifically identification of a germline pathogenic variant in NF1 and not a variant of uncertain significance, is recommended for patients who are suspected of having neurofibromatosis but who do not fulfill clinical criteria. This testing is particularly valuable when clinical features are incomplete in young children or when distinguishing NF1 from phenotypically similar conditions such as Legius syndrome. Testing can include sequence analysis of genomic DNA and/or complementary DNA with gene-targeted deletion analysis. (See table .)
Diagnosing Neurofibromatosis
Type | Criteria |
|---|---|
Neurofibromatosis type 1 (NF1) | Diagnosis of NF1 is established if ≥ 2 of the following features are present:
|
NF2-related schwannomatosis (NF2) | One of the following:
OR Major criteria (2 of the following):
OR One major criterion and 2 of the following minor criteria:
|
Non-NF2 schwannomatosis (schwannomatosis) | SMARCB1- and LZTR1-related schwannomatosis (1 of the following):
22q-related schwannomatosis (all of the following):
Schwannomatosis not otherwise specified (both of the following, no genetic testing done):
|
* These criteria can be counted twice (ie, 2 distinct schwannomas count as 2 minor criteria). | |
† Multiple meningiomas qualify as major criteria. | |
‡ These criteria can be counted only once (ie, bilateral cortical cataracts count as 1 minor criterion). | |
Data from Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med. 2021;23(8):1506-1513. doi:10.1038/s41436-021-01170-5, Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24(9):1967-1977. doi:10.1016/j.gim.2022.05.007, and from Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; October 2, 1998. | |
Diagnosis references
1. Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med. 2021;23(8):1506-1513. doi:10.1038/s41436-021-01170-5
2. Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24(9):1967-1977. doi:10.1016/j.gim.2022.05.007
3. Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; October 2, 1998.
Treatment of Neurofibromatosis
For symptomatic neurofibromas in NF1, surgical removal or targeted medications (eg, MEK inhibitors) for inoperable tumors
For malignant tumors, chemotherapy
For vestibular schwannomas, surgery and bevacizumab in selected cases
For non-NF2 schwannomatosis, primarily pain management
The management of neurofibromatosis is typically multidisciplinary, involving neurology, ophthalmology, orthopedics, and oncology (1). Individualized treatment depends on the specific type (NF1, NF2, or non-NF2 schwannomatosis), the clinical manifestations, and disease severity (2).
In patients with NF1, discrete neurofibromas (cutaneous or plexiform) that cause severe symptoms may require surgical removal or, if small, removal by laser or electrocautery. Because of their depth, surgical removal of plexiform neurofibromas may obliterate function of the involved nerve, and these neurofibromas have a tendency to recur at the site of removal. For symptomatic but inoperable NF1 plexiform neurofibromas, selective mitogen-activated protein kinase (MEK) inhibitors are considered the standard of care and include selumetinib (for patients ≥ 1 year) and mirdametinib (for patients ≥ 2 years). These medications can help reduce the volume of tumors and associated pain and can improve quality of life (3, 4).
Most optic gliomas are asymptomatic. In patients with confirmed neurofibromatosis, clinical monitoring for the development of optic gliomas is advised every 6 to 12 months until the age of 8 (5); thereafter, annual screening is recommended (6). For both progressive optic gliomas and central nervous system lesions that have become malignant, chemotherapy is the treatment of choice. Emerging therapies, particularly other MEK inhibitors (eg, trametinib, binimetinib), tyrosine kinase inhibitors (eg, cabozantinib), and vascular endothelial growth factor inhibitors (eg, bevacizumab), are under investigation as treatment for progressive or refractory disease (7, 8).
The treatment of vestibular schwannomas is primarily surgical. However, slow-growing tumors may not require immediate surgical intervention. In rapidly growing vestibular schwannomas, bevacizumab has shown promising benefit in delaying progression of hearing loss and need for surgery (9, 10). Hearing preservation and augmentation are equally important in the optimal care of these patients; hence all patients with NF2-related schwannomatosis should be referred to an audiologist.
The treatment of non-NF2 schwannomatosis is primarily symptomatic with long-term pain management, including analgesic medications, physical therapy, and nerve blocks. Surgical resection of schwannomas is recommended if the patient has uncontrolled pain or if the schwannomas cause neurologic deficit. Ideally, such patients are cared for by a multidisciplinary team with expertise in the various manifestations of the condition.
Genetic counseling is advisable for all types of neurofibromatosis. If either parent has neurofibromatosis, the risk of their children developing it is 50%; if neither has it, risk for subsequent children is unclear because new mutations are common, particularly in NF1.
Treatment references
1. Miller DT, Freedenberg D, Schorry E, et al. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019;143(5):e20190660. doi:10.1542/peds.2019-0660
2. Stewart DR, Korf BR, Nathanson KL, Stevenson DA, Yohay K. Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20(7):671-682. doi:10.1038/gim.2018.28
3. Chen AP, Coyne GO, Wolters PL, et al. Efficacy and safety of selumetinib in adults with neurofibromatosis type 1 and symptomatic, inoperable plexiform neurofibromas (KOMET): a multicentre, international, randomised, placebo-controlled, parallel, double-blind, phase 3 study. Lancet. 2025;405(10496):2217-2230. doi:10.1016/S0140-6736(25)00986-9
4. Moertel CL, Hirbe AC, Shuhaiber HH, et al. ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma. J Clin Oncol. 2025;43(6):716-729. doi:10.1200/JCO.24.01034
5. Evans DGR, Salvador H, Chang VY, et al. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 1. Clin Cancer Res. 2017;23(12):e46-e53. doi:10.1158/1078-0432.CCR-17-0589
6. Carton C, Evans DG, Blanco I, et al. ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. EClinicalMedicine. 2023;56:101818. Published 2023 Jan 13. doi:10.1016/j.eclinm.2022.101818
7. Amato A, Imbimbo BP, Falsini B. Neurofibromatosis type 1-associated optic pathway gliomas: pathogenesis and emerging treatments. Eur Rev Med Pharmacol Sci. 2023;27(12):5636-5653. doi:10.26355/eurrev_202306_32804
8. Armstrong AE, Belzberg AJ, Crawford JR, Hirbe AC, Wang ZJ. Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas. BMC Cancer. 2023;23(1):553. Published 2023 Jun 16. doi:10.1186/s12885-023-10996-y
9. Van Gompel JJ, Agazzi S, Carlson ML, et al. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on Emerging Therapies for the Treatment of Patients With Vestibular Schwannomas. Neurosurgery. 2018; 82(2):E52-E54. doi:10.1093/neuros/nyx516
10. Screnci M, Puechmaille M, Berton Q, Khalil T, Mom T, Coll G. Bevacizumab for Vestibular Schwannomas in Neurofibromatosis Type 2: A Systematic Review of Tumor Control and Hearing Preservation. J Clin Med. 2024;13(23):7488. Published 2024 Dec 9. doi:10.3390/jcm13237488
Key Points
There are 3 types of neurofibromatosis (NF): NF1, NF2-related schwannomatosis (NF2), and non-NF2 schwannomatosis (schwannomatosis), caused by gene mutations.
NF1 typically causes cutaneous, neurologic, and bone abnormalities but can affect almost any part of the body.
NF2 causes bilateral vestibular schwannomas.
Schwannomatosis causes multiple nonintradermal schwannomas; it does not cause vestibular schwannomas.
Diagnosis is made using clinical criteria; neuroimaging is done if patients have neurologic abnormalities.
Genetic testing is recommended for patients who are suspected to have neurofibromatosis but who do not fulfill clinical criteria.
Neurofibromas that cause severe symptoms may be removed surgically.
Malignant tumors may require chemotherapy.
Kinase inhibitors or vascular endothelial growth factor inhibitors may reduce tumor volume and improve patient quality of life.
Vestibular schwannomas may be removed surgically; rapidly growing schwannomas can be treated with bevacizumab (a vascular endothelial growth factor inhibitor) in some patients.
Non-NF2 schwannomatosis requires symptomatic treatment with long-term pain management.
Drug Information for the Topic





